Created by Jiří Kofránek
1. kapitola z učebnice E.Nečas Patologická fyziologie orgánových systémů ćást 1, Karolinum, 2003 Tento text můžete pouze komentovat - udělejte novou komentářovou buňku s komentovaným textem.
Nehodginské lymfomy (NHL) jsou velice heterogenní skupinou onemocnění vznikajících z různých vývojových stádií B či T lymfocytů.
!!!začátek petitu!!!
Současná klasifikace dle Světové zdravotnické organizace z roku 2022 rozeznává několik desítek typů NHL, které se liší genetickými změnami, histologicky, imunofenotypem, klinickým chováním (agresivní vs indolentní, zohlednění primární lokalizace lymfomu, atd.), atd. NHL odvozené od B lymfocytů jsou častější než NHL odvozené od T lymfocytů (ty tvoří cca 6% všech typů lymfomů). Nejčastější podtypy B lymfomů jsou uvedeny v tabulce 1.8 spolu s častými genetickými změnami a dalšími základními charakteristikami.
!!!konec petitu!!!
Blíže jsou popsány dva podtypy nehodgkinských lymfomů – difúzní velkobuněčný B lymfom (nejčastější agresivní lymfom) a folikulární lymfom (nejčastější indolentní lymfom).
Difúzní velkobuněčný B-lymfom (DLBCL) je agresivní lymfoproliferace odvozená od periferních/zralých B lymfocytů. Jedná se o biologicky heterogenní onemocnění se dvěma genetickými podtypy.
!!!začátek petitu!!!
Geny zvýšeně exprimované jednotlivými podtypy DLBCL korespondují s expresním profilem těch vývojových stadií B lymfocytů, na jejichž úrovni došlo k maligní transformaci. Rozlišujeme tak DLBCL analogický buňkám zárodečných center (GCB, germinal centre B cell-like) nebo postgerminálním aktivovaným B buňkám (ABC, activated B cell-like). V patogenezi těchto dvou hlavních podtypů DLBCL se uplatňují odlišné onkogenní mechanismy a tyto lymfomy mají odlišný profil genetických změn (tabulka 1.8). Určením podtypu DLBCL lze predikovat prognózu pacientů, která je významně lepší pro GCB podtyp DLBCL ve srovnání s ABC DLBCL.
!!!konec petitu!!!
V klinickém obraze bývají často přítomny celkové příznaky, tzv. B symptomy (horečka, váhový úbytek, noční poty) a lymfadenopatie. Časté je také postižení extranodálních orgánů či tkání (např. gastrointestinální trakt, plíce, ledviny, centrální nervový systém, atd.). U DLBCL platí, že může být infiltrován téměř jakýkoliv orgán či tkáň. Symptomy jsou pak velmi různorodé a odvozené od postižení jednotlivých orgánů či tkání. Kostní dřen bývá infiltrována u menšiny pacientů a leukemizace (tj. vyplavování nádorových buněk do periferní krve) je vzácná.
Folikulární lymfom (FL) je indolentní lymfoproliferace odvozená od periferních/zralých B lymfocytů. U naprosté většiny případů FL je detekována chromozomální translokace t(14;18), která vede ke zvýšené expresi anti-apoptotického proteinu BCL2 a prodlouženému přežívání B lymfocytů. Chromozomální translokace t(14;18) je pravděpodobně iniciální onkogenní změnou, ale sama o sobě není dostačující a pro vznik FL je nutná akumulace dalších genetických změn.
Pro FL je typická generalizovaná lymfadenopatie, která zpočátku nemusí způsobovat žádné větší obtíže. Častá je také infiltrace kostní dřeně nádorovými buňkami, která se vyskytuje u více než 50 % pacientů s FL. Postižení extranodálních orgánů či tkání je u FL méně časté. U tohoto podtypu lymfomu lze zpočátku zvolit strategii watch and wait (pouze sledování onemocnění) a zahájit léčbu v době, kdy je přítomna velká nádorová masa projevující se klinickými příznaky nebo laboratorními známkami progrese onemocnění (např. velká nádorová masa způsobující útlak důležitých orgánů/tkání, cytopenie v důsledku narůstající infiltrace kostní dřeně, významné výpotky, atd.). FL se může transformovat do agresivního lymfomu, nejčastěji do difúzního velkobuněčného B-lymfomu.
Chronická lymfocytární leukémie (CLL) patří mezi indolentní lymfoproliferace vznikající ze zralých B lymfocytů. Pro CLL je typická postupná akumulace monoklonálních B lymfocytů, které mají odolnost vůči apoptóze. Nádorové lymfocyty se akumulují v kostní dřeni, periferní krvi, lymfatických uzlinách, slezině a játrech. Zvýšená odolnost vůči apoptóze je u CLL způsobena zvýšenou expresí antiapoptotických proteinů BCL2 a MCL1.
!!!začátek petitu!!!
U části případů CLL bývá detekována delece chromozomální oblasti 13q14, kde jsou lokalizovány geny kódující inhibiční microRNA (miR15 a miR16), které za normálních okolností snižují expresi genu BCL2.
!!!konec petitu!!!
Pro CLL je typická lymfocytóza v periferní krvi a infiltrace kostní dřeně. Celkový počet leukocytů se může zvýšit na 10 x 109/l krve, ale může také dosáhnout hodnot až 200 x 109/l krve. Onemocnění může být zpočátku zcela asymptomatické a může být diagnostikováno náhodně při vyšetření krevního obrazu. Akumulace nádorových lymfocytů v kostní dřeni může způsobit útlak normální krvetvorby s výslednou anémií a trombocytopénií a pro ně typickými příznaky. Častá bývá také lymfadenopatie, splenomegalie či hepatomegalie. Přestože je množství B-lymfocytů v periferní krvi a v kostní dřeni zvýšené, bývá často snížená koncentrace imunoglobulinů v plazmě, což se podílí na častějších infekcích. U části nemocných s CLL mohou být projevy autoimunitního poškození, např. protilátkami proti erytrocytům (s výslednou autoimunitní hemolytickou anémií) nebo trombocytům (s výslednou imunní trombocytopénií). CLL se může transformovat do agresivní lymfoproliferace, nejčastěji do difúzního velkobuněčného B-lymfomu.
Hodgkinův lymfom (HL) se vyčleňuje od ostatních lymfomů jednak z historického důvodu (byl popsán v roce 1832 Thomasem Hodgkinem), jednak vzhledem k několika unikátním biologickým a klinickým rysům tohoto onemocnění.
Pro HL je typická přítomnost velkých nádorových lymfomových buněk – buněk Reedové-Sternberga (RS). Ačkoli nebyl dlouho znám fyziologický protějšek RS buněk, molekulárně biologickými metodami bylo prokázáno, že HL vzniká z B lymfocytů zárodečného centra (nachází se v lymfatické uzlině) a lze jej tedy řadit mezi periferní/zralé B lymfoproliferace. RS buňky typicky tvoří méně než 1% všech buněk tumoru a jsou obklopeny zánětlivými buňkami (B a T lymfocyty, granulocyty, makrofágy, fibroblasty), které tvoří tzv. nádorové mikroprostředí. Mezi RS buňkami a buňkami nádorového mikroprostředí dochází k vzájemným interakcím ať už prostřednictvím produkce cytokinů či vzájemnou vazbou pomocí povrchových proteinů. Buňky nádorového mikroprostředí produkují řadu cytokinů (např. IFNγ, IL-2, IL-10 a IL-13), které zajišťují růst a prodloužené přežívání RS buněk. Z terapeutického hlediska je velmi důležitá interakce PD-1 ligandu na povrchu RS buněk a PD-1 receptoru na povrchu T lymocytů. Tato interakce působí na T lymfocyty inhibičně, což vede k neefektivní protinádorové imunitě.
Přestože je HL onemocněním, které vzniká z B-lymfocytů, mají RS buňky specifický imunofenotyp s expresí některých znaků T lymfocytů (CD30+) a často s chyběním typických znaků B lymfocytů.
!!!začátek petitu!!!
HL postihuje typicky mladé dospělé lidi nebo lidi kolem 60. roku věku. HL se vyskytuje mnohonásobně častěji u jednovaječných dvojčat. V některých případech Hodgkinovy nemoci byla prokázána infekce virem EBV. Stejně jako u nehodgkinských lymfomů zvyšuje výskyt Hodgkinovy nemoci dlouhodobá imunosupresivní léčba, imunosuprese spojená s infekcí virem HIV a autoimunitní onemocnění.
!!!konec petitu!!!
Klinicky se jedná o agresivní lymfoproliferaci. V klinickém obraze dominuje lymfadenopatie (často krční, mediastinální) a přítomnost B symptomů (horečky, hubnutí a noční poty, kašel). U části pacientů může mediastinální masa způsobovat syndrom horní duté žíly, který vzniká útlakem horní duté žíly nádorovou masou a projevuje se otokem obličeje, krku, horních končetin a dilatací povrchových žil. U části pacientů s HL se může objevit také svědění kůže bez přítomnosti exantému. Infiltrace kostní dřeně je u HL velmi vzácná.
Onemocnění je chemo- a radiosenzitivní a má velmi dobrou prognózu.
Obrázek 1.8 Podrobnosti viz text
Akutní lymfoblastová leukémie (ALL) a lymfoblastový lymfom (LL) patří mezi prekurzorové lymfoproliferace a vznikají maligní transformací lymfoidní progenitorové buňky. Jedná se klinicky o agresivní onemocnění s rychlým nástupem příznaků. ALL a LL sdílí řadu biologických charakteristik, ale liší se zejména klinickými symptomy (symptomy související s infiltrací kostní dřeně u ALL vs. symptomy doprovázející lymfomovou nádorovou masu u LL). Jejich jednoznačné odlišení je pak založeno na míře infiltrace kostní dřeně. Pokud infiltrace kostní dřeně nádorovými buňkami přesahuje 25 %, onemocnění je klasifikováno jako ALL.
Pro tato onemocnění je typická akumulace nezralých patologických lymfoblastů v kostní dřeni, periferní krvi, lymfatických uzlinách, slezině, játrech. Může být přítomna také infiltrace extralymfatických orgánů a tkání, typicky centrálního nervového systému, varlat či ovárií. U ALL patologický klon utlačuje normální krvetvorbu a vede k anémii, trombocytopenii a neutropenii. Celkový počet leukocytů v periferní krvi může být zvýšený, normální či snížený, důležitý je diferenciální rozpočet leukocytů s průkazem nezralých buněk - blastů v periferní krvi. Mezi typické příznaky ALL patrří zejména únava (v důsledku anémie), krvácivé projevy (při trombocytopenii), horečka a časté infekce (v důsledku neutropenie) a bolesti kostí, typicky dolních končetin (při expanzi nádorového klonu v kostní dřeni).
Prekurzorové lymfoproliferace lze dělit na B-ALL/B-LL či T-ALL/T-LL, podle toho zda vykazují imunofenotyp B či T lymfocytů. B-ALL tvoří cca 85 % všech prekurzorových lymfoproliferací.
!!!začátek petitu!!!
Současná WHO klasifikace odráží patogenezi těchto onemocnění a dále dělí B-ALL/LL do skupin dle přítomnosti či nepřítomnosti určitých genetických abnormalit na:
B-ALL/B-LL blíže nespecifikovaná
B-ALL/B-LL s rekurentními cytogenetickými změnami
Mezi genetické změny rekurentně nacházené u B-ALL/LL patří např. chromozomální translokace t(9;22) vedoucí ke vzniku fúzního genu BCR-ABL a fúzního proteinu BCR-ABL, který má konstitutivně aktivní tyrozinkinázovou aktivitu a vede k zvýšené proliferaci nádorového klonu. Další rekurentně se vyskytující chromozomální translokací je t(12;21), která vede k fúzi genů ETV6-RUNX1. Fúzní protein ETV6-RUNX1 zahrnuje dva transkripční faktory a ovlivňuje celou řadu intracelulárních signálních drah, jejichž deregulace má za následek např. inhibici apoptózy, prodloužené přežívání, zvýšení proliferace, atd. U ALL jsou také časté změny počtu chromozomů jako je hyperdiploidie nebo hypodiploidie.
!!!konec petitu!!!
Přítomnost či nepřítomnost určitých cytogenetických abnormalit má významné prognostické hledisko a určuje intenzitu a typ terapie pacientů s ALL.
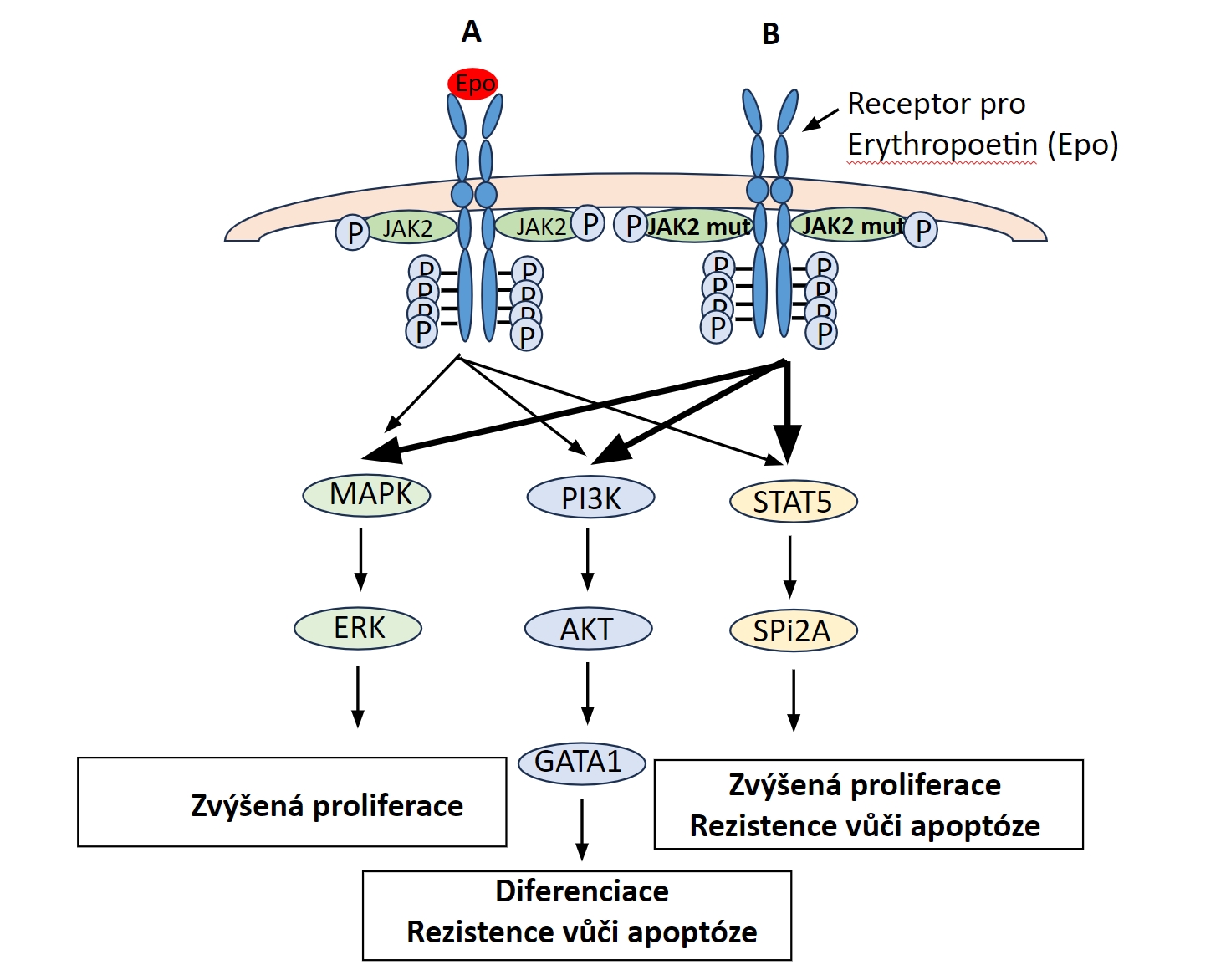
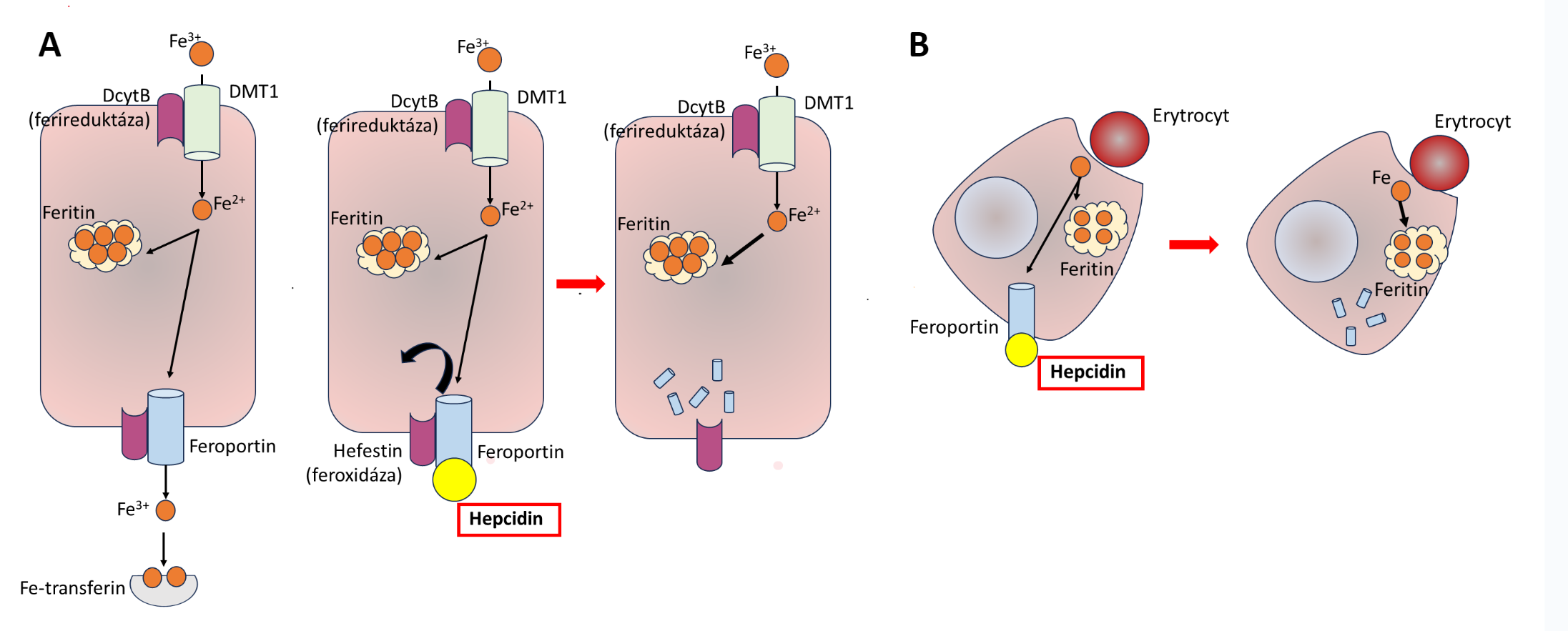
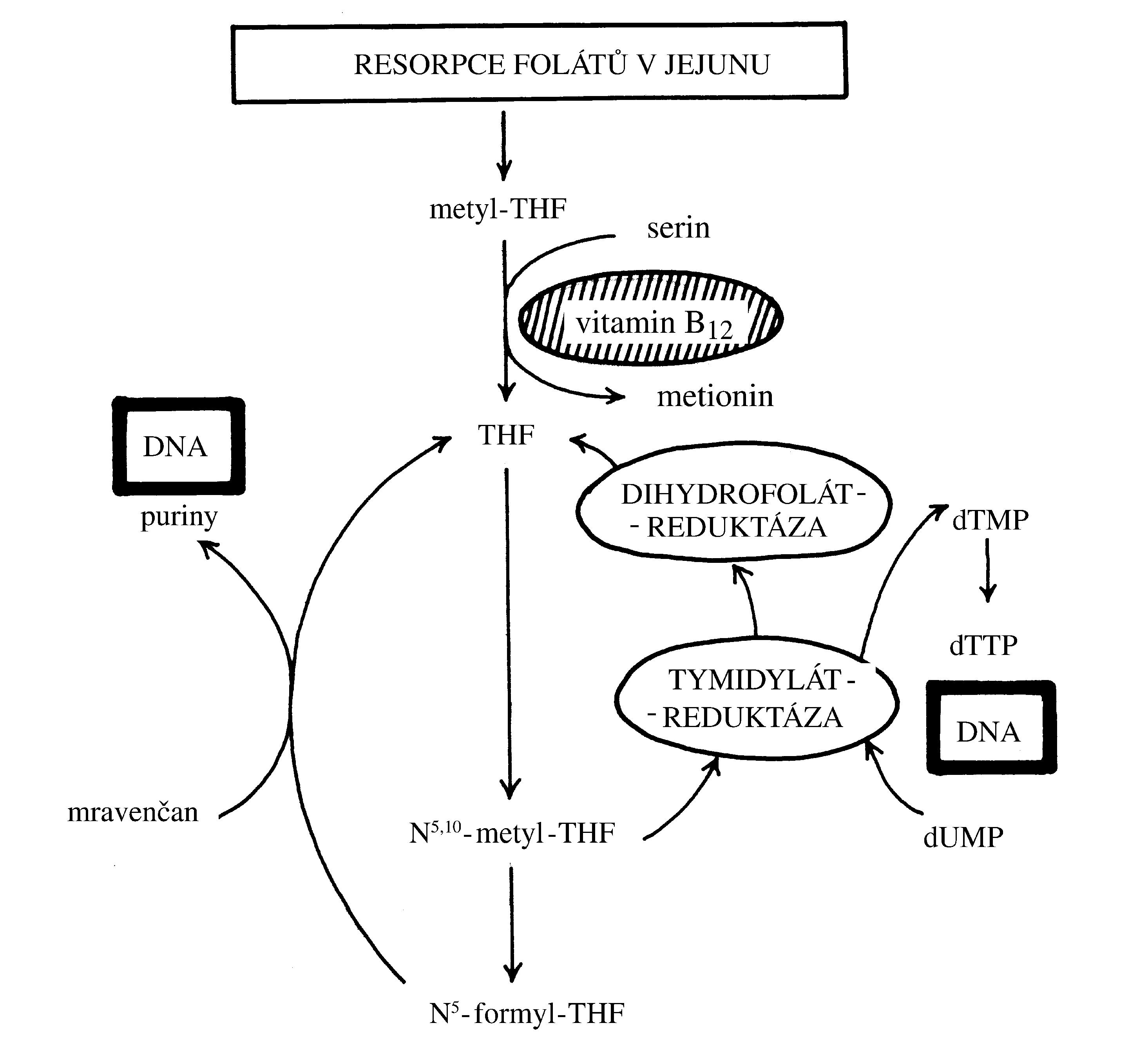
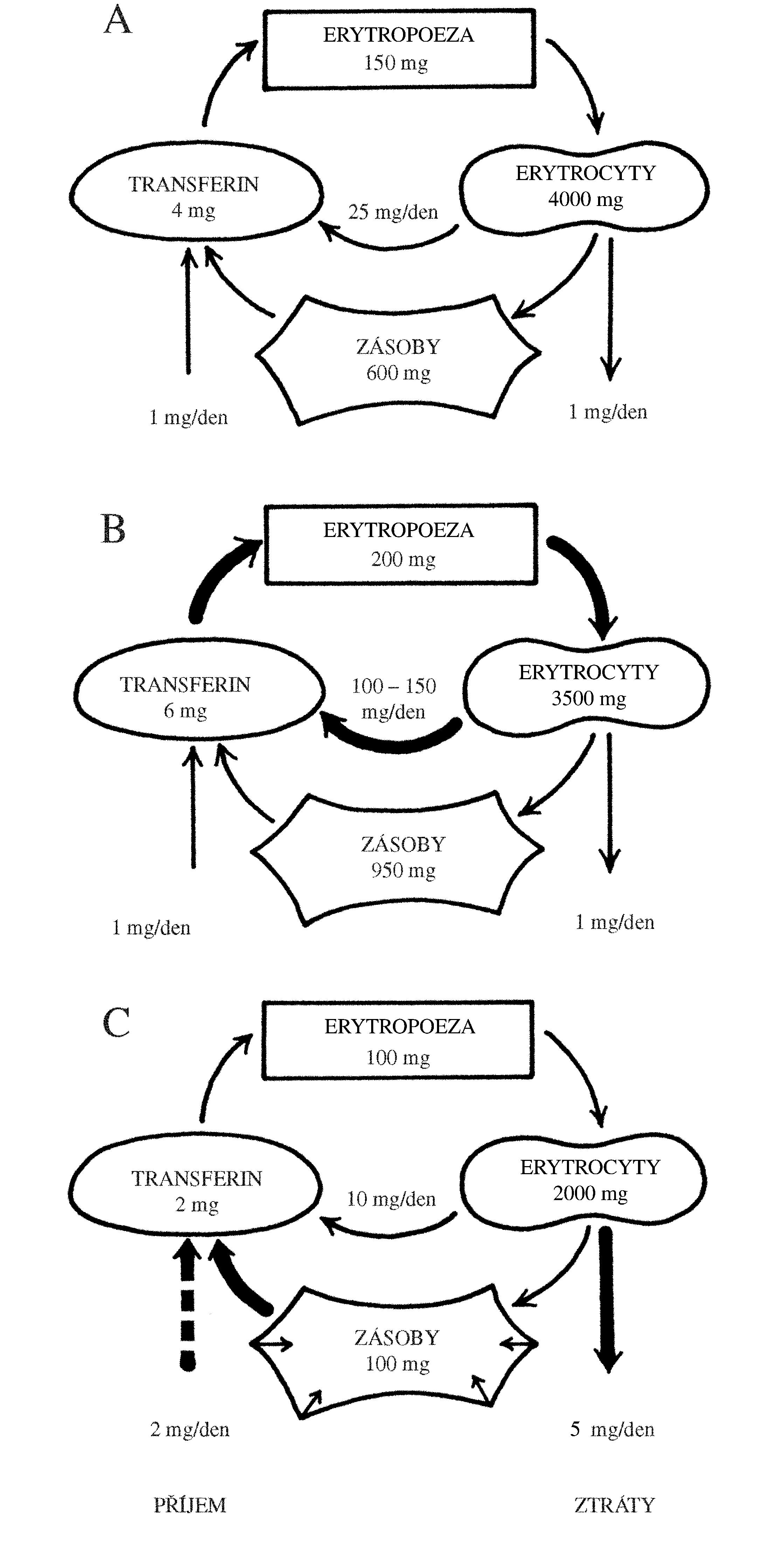
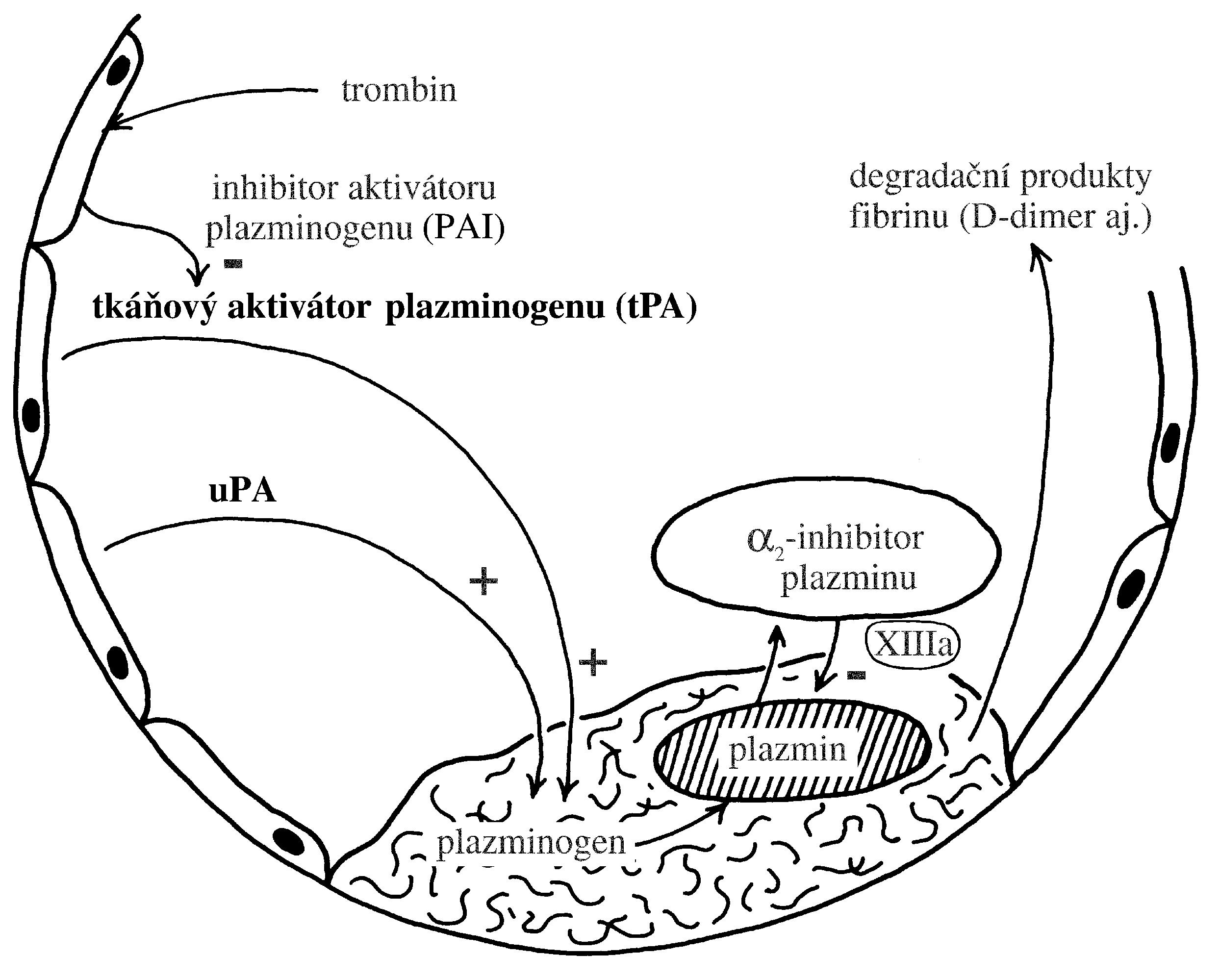
Polycythaemia vera (PV), „pravá polycytémie“, patří mezi získané7 myeloproliferativní onemocnění s monoklonální krvetvorbou odvozenou od patologického klonu. Molekulární podstatou PV je aktivační bodová mutace v genu JAK2 (V617F nebo Val617Phe, bodová mutace způsobující záměnu aminokyseliny valinu za fenylalanin v pozici 617), který kóduje intracelulární tyrozinkinázu, která spolupůsobí s povrchovými buněčnými receptory pro růstové faktory – receptorem pro erythropoetin, receptorem pro trombopoetin a receptorem pro G-CSF. Mutace genu JAK2 (V617F) je u naprosté většiny případů PV (96 %), ale vyskytuje se i u dalších myeloproliferativních onemocnění – např. u esenciální trombocytémie (50 % případů) či primární myelofibrózy (50 % případů). Mutovaná JAK2 kináza konstitutivně aktivuje signální dráhu JAK/STAT5 (a další intracelulární signální dráhy), což vede k transkripci genů způsobujících zvýšení proliferace a rezistenci vůči apoptóze (Obr. 1.6). Výsledkem je trvalé zvýšení erytropoézy, která je nezávislá na stimulaci erythropoetinem, jehož hladina je nízká, nižší než je jeho hladina u zdravých jedinců (Obr. 1.8)8. Nízká hladina erythropoetinu umožňuje odlišit od PV stavy doprovázené sekundární polycytémií (např. v důsledku chronické hypoxie), při nichž je hladina erythropoetinu v krvi zvýšená. Snížení závislosti erytroidní diferenciace na erytropoetinu způsobuje nadměrnou tvorbu erytrocytů, jejichž celkové množství v krvi se zvýší (nad 32/36 ml erytromasy/kg t. hm. u žen/mužů), čímž vzniká obraz polycytémie, tj. zvýšení koncentrace hemoglobinu, hematokritu a počtu erytrocytů v krvi. V periferní krvi bývá dále zvýšený počet granulocytů a trombocytů. Jejich zvýšení svědčí pro to, že genetická změna u PV vznikla na úrovni myeloidní progenitorové buňky. Zvýšení počtu granulocytů a trombocytů však není tak výrazné jako v případě erytrocytů.
!!!obr. 1.8.!!!
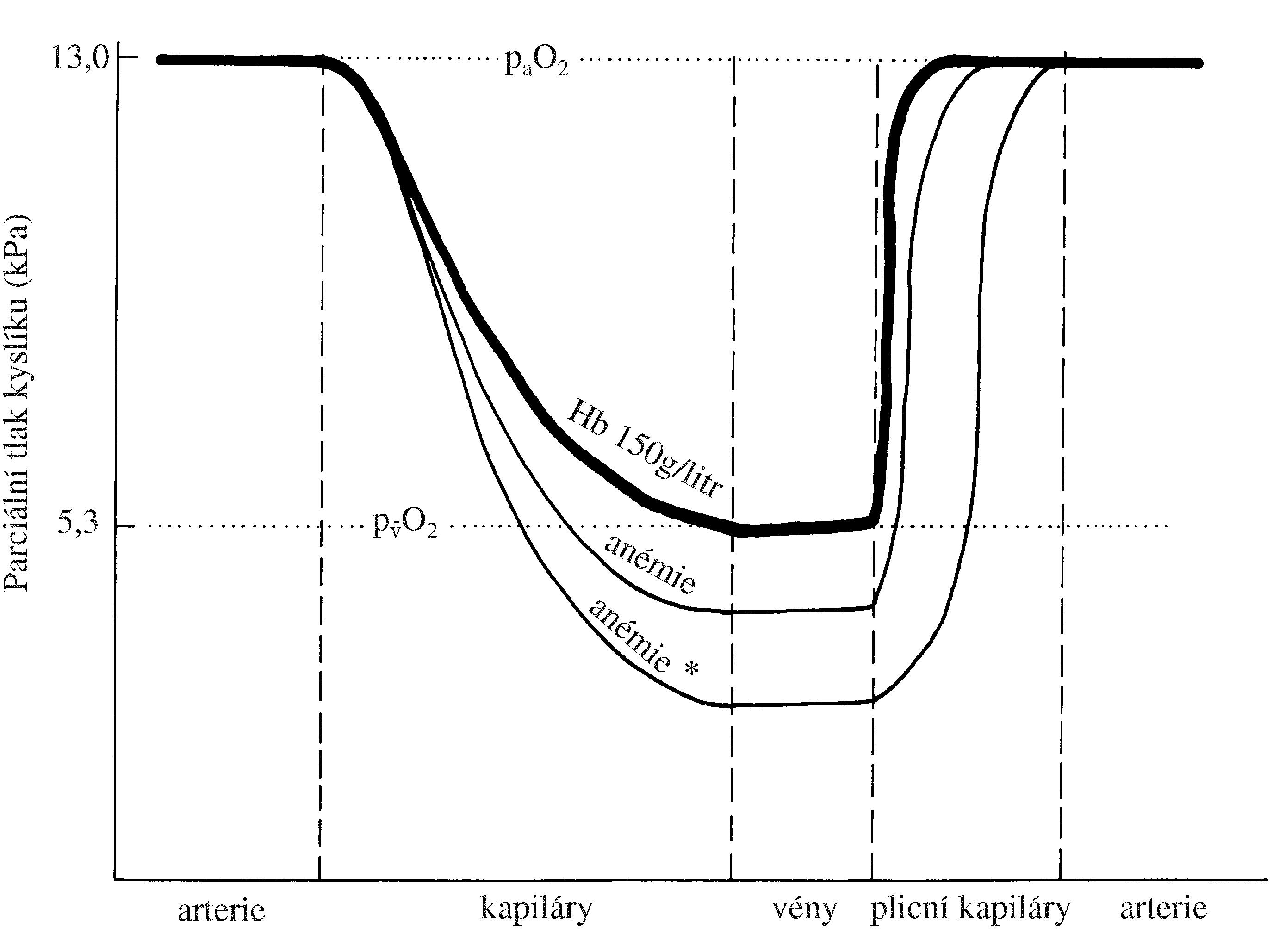
Většina klinických projevů u PV vzniká v důsledu zvýšení hematokritu a hypervolémie. Hypervolémie, většinou provázená zvýšeným hematokritem, působí zvýšení krevního tlaku, které je kompenzováno vazodilatací v cévním řečišti (příznakem je např. teplá, dobře prokrvená kůže). Dále bývají přítomny trombotické či krvácivé komplikace, které jsou nejčastější příčinou úmrtí u pacientů s PV. Trombózy bývají častěji arteriální než venózní. Jejich příčina je multifaktoriální, podílí se na ní hyperviskozita krve a s ní související zpomalení jejího toku v cirkulaci, porucha funkce trombocytů a aktivace granulocytů. Může docházet i ke krvácivým projevům, jejichž intenzitu zvyšuje hypervolémie. Mezi další krvácivé projevy patří epistaxe, krvácení do gastrointestinálního a urogenitálního traktu a prodloužené krvácení po poranění. Komplikací může být závažné krvácení do CNS. V důsledku zvýšené viskozity krve a jejího zpomaleného toku v cirkulaci se zvyšuje srdeční práce, kůže a sliznice mohou snadněji nabýt cyanotického zabarvení, protože je zvýšené množství redukovaného (deoxygenovaného) hemoglobinu v kapilárách (saturace hemoglobinu kyslíkem v arteriální krvi je však prakticky normální, přestože hodnota paO2 může být lehce snížená).
Z dlouhodobého hlediska může PV transformovat do akutní myeloidní leukémie nebo se může vyvinout fibróza kostní dřeně neodlišitelná od primární myelofibrózy. Pro fázi myelofibrózy ja pak typická extramedulární hematopoéza ve slezině, která způsobuje splenomegalii.
7Vzácně existují rodiny s mnohočetným výskytem pravé polycytémie. Ve srovnání s PV mají familiární polycytémie odlišnou molekulární podstatu.
Monoklonální gamapatie nejistého významu (MGUS) je charakteristická přítomností klonálních plazmatických buněk v kostní dřeni a přítomností paraproteinu, bez dalších laboratorních abnormalit a bez známek poškození orgánů či tkání produkovaným paraproteinem. MGUS je iniciálním stadiem malignit odvozených od plazmatických buněk a typicky předchází diagnózu mnohočetného myelomu. Bývá náhodným laboratorním nálezem.
Tabulka 1.5. WHO klasifikace akutní myeloidní leukémie (AML)
| AML se specifickou genetickou abnormalitou |
| Akutní promyelocytární leukémie s fúzí genů PML::RARA |
| AML s fúzí genů AML1::ETO (RUNX1::RUNX1T) |
| AML s fúzí genů CBFB::MYH11 |
| AML s fúzí genů DEK::NUP214 |
| AML s fúzí genů RBM15::MRTFA |
| AML s fúzí genů BCR::ABL1 |
| AML s přestavbou genu KMT2A (MLL1) |
| AML s přestavbou genu MECOM |
| AML s přestavbou genu NUP98 |
| AML s mutací genu NPM1 |
| AML s mutací genu CEBPA |
| AML asociovaná s myelodysplázií |
| AML s jinou definovanou genetickou abnormalitou |
| AML definovaná stupněm diferenciace |
| AML s minimální diferenciací |
| AML bez maturace |
| AML s maturací |
| Akutní bazofilní leukémie |
| Akutní myelomonocytární leukémie |
| Akutní monocytární leukémie |
| Akutní erytroleukémie |
| Akutní megakaryoblastová leukémie |
8Normální hladina erytropoetinu je 10–20 jednotek/l plazmy. Protože současné metody jeho stanovení založené na principu ELISA nebo RIA mají spodní detekční citlivost okolo 3 jednotek/litr, potvrzují diagnózu pravé polycytémie hodnoty, které nejsou vyšší než normální.
Esenciální trombocytémie (ET) patří mezi myeloproliferativní onemocnění, jehož hlavním znakem je zvýšení počtu trombocytů v krvi nad 450 x 109/l. ET je asociována s genetickými změnami, které vznikají na úrovni myeloidní progenitorové buňky nebo hematopoetické kmenové buňky a způsobují patologickou monoklonální krvetvorbou. Aktivační bodová mutace genu JAK2 (V617F) je u asi 50 % případů. V ostatních případech (20–40 %) je mutace genu CALR (kóduje protein calretikulin) nebo aktivační mutace genu MPL (5-10 %; kóduje receptor pro trombopoetin).
!!!začátek petitu!!!
Calreticulin se nachází v endoplazmatickém retikulu a patří mezi chaperonové proteiny, jejichž funkcí je zajistit správné sbalování nově vzniklých proteinů v endoplazmatickém retikulu. Mutovaná forma calretikulinu se váže na protein MPL a aktivuje ho. Všechny popsané genetické změny vedou k aktivaci signální dráhy JAK/STAT, což vede k transkripci genů způsobujících buněčnou proliferaci. Výsledkem je na růstových faktorech nezávislá trombopoéza.
!!!konec petitu!!!
V kostní dřeni je zvýšený výskyt megakaryoblastů a megakaryocytů, v periferní krvi je zvýšený počet trombocytů (v periferní krvi někdy mohou být nalézány abnormálně velké trombocyty). V klinických projevech dominuje zvýšený výskyt trombóz či krvácivých projevů (ty vznikají v důsledku nedostatečné funkce trombocytů), podobně jako u PV. ET vzácně přechází do akutní myeloidní leukémie či myelofibrózy.
Při podezření na ET je zejména důležité odlišit stavy doprovázené sekundárním zvýšením počtu trombocytů – např. infekce, zánět, stavy spojené s krvácením, poškozením tkání, infarkt myokardu, nádory, stavy po splenektomii atd. Reaktivní zvýšení trombocytů bývá u zánětlivých stavů způsobeno zvýšenou hladinou prozánětových cytokinů (IL-1b, IL-6, IL-11).
Tab. K1.1 Hematologické vyšetření
| Den hospitalizace | |||||||
| 1. | 3. | 5. | 10. | 20. | 31. | 42. | |
| Hemoglobin (g/l) | 87 | 67 | 82 | 94 | 66 | 72 | 50 |
| Erytrocyty (x 1012/l) | 2,8 | 1,9 | 2,5 | 2,7 | 2,0 | 2,3 | 1,5 |
| Hematokrit (%) | 26 | 19 | 23 | 26 | 18 | 21 | 15 |
| MCV (fl) | 92 | 103 | 95 | 98 | 94 | 91 | 97 |
| MCH (pg) | 31 | 35 | 32 | 24* | 33 | 31 | 33 |
| Retikulocyty (%) | 0,2 | 0,1 | |||||
| Leukocyty (x 109/l) | 2,0 | 1,2 | 1,2 | 2,8 | 1,0 | 0,8 | 0,2 |
| Neutrofilní granulocyty (%) | 9 | 5 | 2 | 0 | |||
| Tyčky (%) | 3 | 0 | 0 | 0 | |||
| Eozinofilní granulocyty (%) | 0 | 0 | 0 | 0 | |||
| Bazofilní granulocyty (%) | 0 | 0 | 0 | 0 | |||
| Monocyty (%) | 3 | 1 | 5 | 4 | |||
| Lymfocyty (%) | 69 | 94 | 92 | 96 | |||
| Blasty (%) | 15 | 0 | 0 | 0 | |||
| Trombocyty (x 109/l) | 7 | 9 | 23 | 8 | 19 | 3 | 16 |
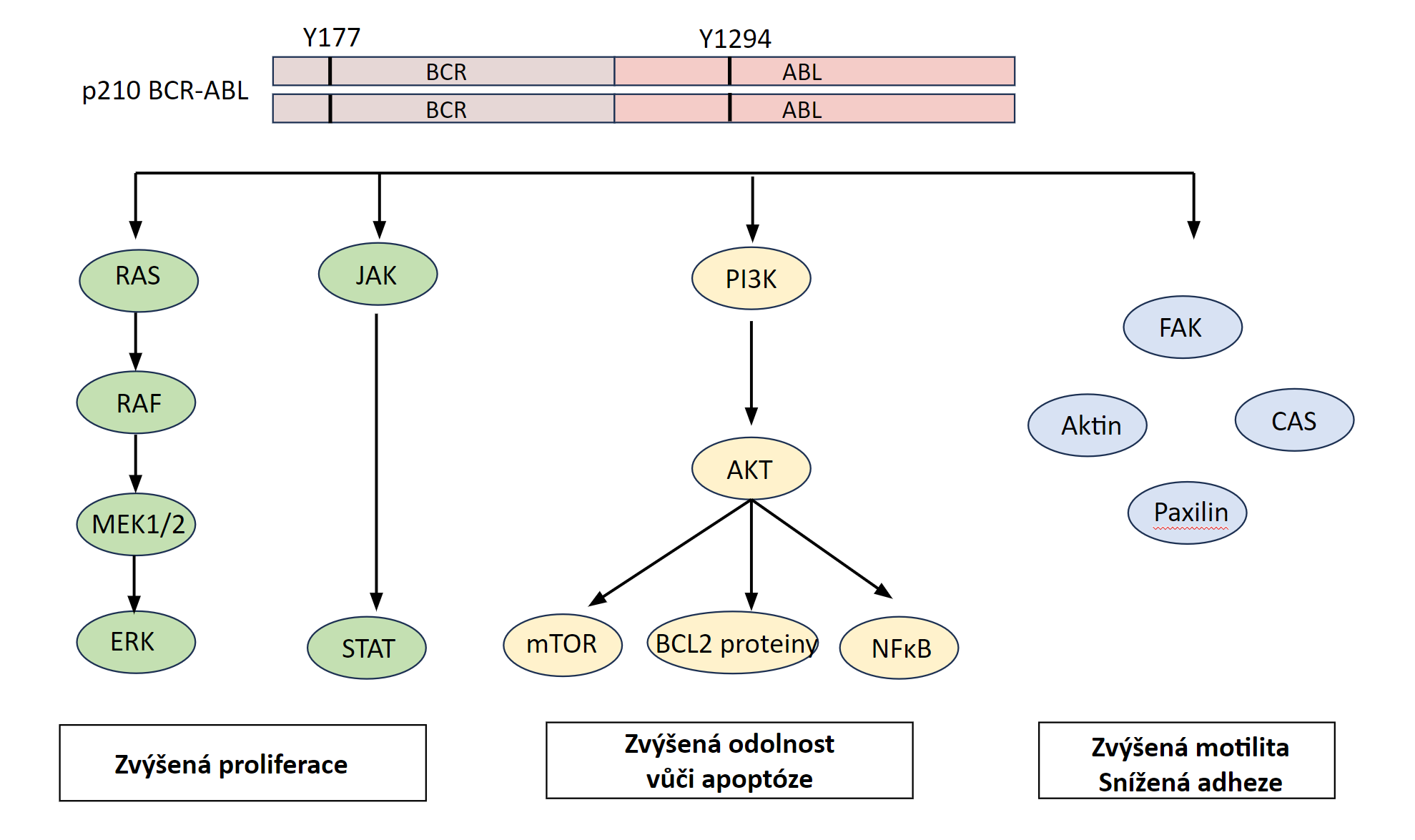
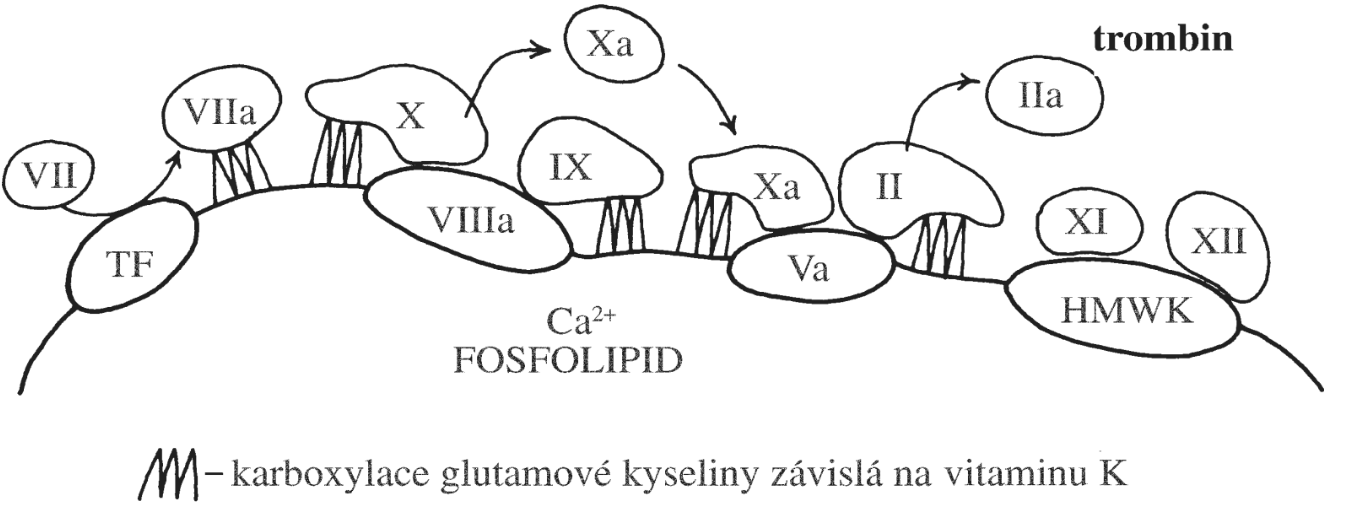
Chronická myeloidní leukémie (CML) patří mezi myeloproliferativní onemocnění a vzniká v důsledku genetické změny postihující multipotentní hematopoetickou kmenovou buňku. CML je charakterizována přítomností chromozomální translokace t(9,22), která dává vznik tzv. philadelphskému chromozomu – Ph-chromozomu. Jedná se o translokací zkrácený chromozom 22. Tato genetická změna bývá u 95 % všech případů CML. Pro vznik a rozvoj CML je podstatná skutečnost, že se při této chromozomální translokaci na 22. chromozomu spojí protoonkogen c-abl (Abelson murine leukemia), který se normálně nachází na dlouhém raménku 9. chromozomu a kóduje thyrosinkinázu, s genem bcr (break cluster region) z dlouhého raménka 22. chromozomu. Fúzní gen bcr/abl, který tím vznikne, je exprimován jako funkčně abnormální cytoplazmatický protein p210 BCR-ABL (podle své molekulové hmotnosti 210 kDa). Místo zlomu na chromozomu 22 může být lokalizováno i v jiné pozici a dochází k expresi fúzního proteinu s odlišnou molekulovou hmotností - p190 BCR-ABL nebo p230 BCR-ABL, což jsou méně časté varianty). Patologický protein p210 BCR-ABL má silnou tyrozinkinázovou aktivitu. Důsledkem jeho přítomnosti v buňce je fosforylace řady buněčných proteinů, zahrnujících proteiny cytoskeletu, transkripční faktory v buněčném jádře, mitochondriální proteiny aj. Fosforylovány jsou i jiné tyrozinkinázy, které pak fosforylují další proteiny.
Fosforylace řady buněčných proteinů, jednak přímým účinkem p210 a jednak dalšími aktivovanými tyrozinovými kinázami, způsobí změny postižených buněk (Obr. 1.7). Zvýší se jejich proliferační aktivita (mitogenní účinek), sníží se jejich adhezivita ke stromatu krvetvorné tkáně, zvýší se jejich rezistence k apoptóze a zvýší se nestabilita jejich genomu. Leukemický klon má tendenci získávat další mutace, což zvyšuje jeho maligní potenciáln tohoto klonu a klinicky se projevuje jako transformace do agresivnější podoby nemoci (akcelerovaná a blastická fáze onemocnění). CML může přecházet do AML nebo do akutní lymfoblastové leukémie, což nasvědčuje tomu, že onemocnění vzniká z multipotentní hematopoetické kmenové buňky.
!!!obr. 1.7.!!!
V periferní krvi bývá výrazně zvýšený počet neutrofilních granulocytů a bývají přítomné nezralé prekurzory granulocytů (blasty, promyelocyty, myelocyty, metamyelocyty), může být přítomna eozinofile, bazofilie, monocytóza. Celkový počet leukocytů bývá často výrazně zvýšen (nejednou vyšší než 100 x 109/l krve). Přestože mají leukemické buňky morfologické znaky zralých granulocytů, jsou jejich antibakteriální funkce nedokonalé, což zbůsobuje infekční komplikace. Počet leukocytů může být vzácně natolik zvýšený, že se významně zvyšuje celková viskozita krve, což způsobuje zpomalení až zastavení průtoku krve některými částmi mikrocirkulace, což je stav označovaný jako leukostáza. U části nemocných s CML může být vyšší počet trombocytů. Mohou se vyskytnout jak trombotické komplikace, tak krvácivé projevy, protože trombocyty jsou funkčně abnormální. V krevním obraze bývá často přítomna anémie. Vedle celkových nespecifických příznaků, jakými jsou únava, hubnutí, se mohou přiřadit i poruchy, jejichž zdrojem je místní tkáňová ischémie. Může se jednat o různé neurologické symptomy, poruchy zraku aj. CML může způsobovat výraznou a symptomatickou splenomegalii a hepatomegalii, která je způsobena infiltrací sleziny patologickým klonem buněk či přítomností extramedulární hematopoézy (viz dále).
!!!začátek petitu!!!
Tyrozinkinázová aktivita p210 je pro vznik CML fenotypu esenciální. Významným přelomem v léčbě CML bylo objevení vysoce specifických inhibitorů tyrozinkinázové aktivity p210. I když inhibice účinku patologické tyrozin-kinázy nevede k vyléčení CML, tj. odstranění patologického klonu, naprostá většina pacientů odpoví velmi dobře na tuto terapii a dosahují dlouhodobé remise onemocnění. Část pacientů však vyvine mutace v oblasti fúzního genu BCR-ABL, které jsou asociovány s rezistencí na inhibitor, což se projeví klinicky jako relaps onemocnění. Byly však vyvinuty další inhibitory tyrozinkinázové aktivity p210, které mají odlišné vazebné místo a mohou být použity pro léčbu CML pacientů se získanou rezistencí.
!!!konec petitu!!!
Obrázek 1.7 Podrobnosti viz text v kapitole
Mezi lymfoproliferativní onemocnění odvozené od plazmatických buněk p atří:
Tato onemocnění vznikají maligní transformací zralého B lymfocytu, který je zdrojem imunoglobulinu. Jedná se o klonánlí expanzi abnormálních plazmatických buněk. Buňky secernují imunoglobulin, a je pro tato onemocnění je proto typická přítomnost monoklonální gamapatie, což je označení pro nadměrnou tvorbu patologického imunoglobulinu, tzv. M-komponenty (M od monoklonální) s elektroforetickou pohyblivostí γ-globulinů.
Secernovaná M-komponenta9 může být plnohodnotnou protilátkou kterékoli imunoglobulinové třídy (nejčastěji IgG nebo IgA), nebo může být jen izolovaným těžkým nebo lehkým imunoglobulinovým řetězcem. Bývá též označována jako paraprotein.Množství secernovaných imunoglobulinů může být značné a jejich koncentrace v plazmě odráží do jisté míry velikost patologického klonu. Produkuje-li nádorový klon lehký imunoglobulinový řetězec, přechází tento glomerulární membránou do moče (mol. hmotnost asi 25 kDa), kde je přítomen jako tzv. Bence Jonesova bílkovina. Přítomnost M-komponenty v plazmě může způsobit, při její značné koncentraci, hyperviskózní syndrom.
!!!začátek petitu!!!
Hyperviskózní syndrom hrozí, přesáhne-li koncentrace IgM 40 g/l, IgG3 50 g/l a IgA 70 g/l. Viskozita plazmy se zvyšuje z normální hodnoty 1,8 na hodnoty 5–610. Hyperviskózní syndrom může způsobit poruchy zraku (specificky se projevuje na očním pozadí) a různé neurologické příznaky, zpomalení proudění krve predisponuje ke vzniku trombóz.
!!!konec petitu!!!
Specifické poruchy mikrocirkulace vznikají tehdy, má-li M-komponenta charakter kryoglobulinů. U nemocných se pak může projevit Raynaudův syndrom, což je ischémie akrálních částí těla (prsty, nos, ušní boltce) při jejich prochladnutí.
Tvorba paraproteinu, M-komponenty, je většinou provázena snížením tvorby normálních imunoglobulinů a také jejich zvýšenou degradací. Přestože je v plazmě zvýšená celková koncentrace imunoglobulinů, jsou monoklonální gamapatie typicky provázeny sníženou odolností vůči bakteriálním infekcím.
Mnohočetný myelom (MM) je onemocnění charakterizované:
- přítomností klonálních plazmatických buněk v kostní dřeni (vzácněji extramedulárně)
- přítomností paraproteinu v séru (vzácně existuje MM bez sekrece paraproteinu),
- přítomností hyperkalcémie, renálního selhání, anémie, osteolytických kostních změn
U MM byla popsána řada genetických změn, které se uplatňují v patogenezi tohoto onemocnění. Tyto změny bývají mnohočetné a zahrnují chromozomální translokace, trizomie/monozomie určitých chromozomů, či ztráty určitých chromozomálních oblastí (např. 17p, kde je lokalizován tumor supresorový gen TP53), ale i bodové mutace řady genů (NRAS, KRAS).
!!!začátek petitu!!!
Chromozomální translokace často zahrnující imunoglobulinové geny, zejména IGH lokus na chromozomu 14 a některý protoonkogen např. gen pro cyklin D1 (CCND1) lokalizovaný na dlouhém raménku chromozomu 11, t(11;14), cyklin D3 lokalizovaný na krátkém raménku chromozomu 6, t(6;14), nebo gen kódující receptor pro fibroblastový růstový faktor 3 (FGFR3) a gen kódující specifickou methyltransferázu histonů (MMSET/WHSC1), které jsou lokalizované na krátkém raménku chromozomu 4, t(4;14) nebo gen kódující transkripční faktor MAF, t(14;16).
!!!konec petitu!!!
Patologické buňky, podobné zralým plazmatickým buňkám, se hromadí v kostní dřeni a mohou utlačovat normální krvetvorbu. Pro MM bývá typická normochromní normocytární anémie, na které se vedle infiltrace kostní dřeně nádorovými plazmatickými buňkami může podílet poškození ledvin púaraproteinem.
Vzácněji tvoří plazmocytom solidní nádor lokalizovaný v kosti nebo jinde extramedulárně. U MM dochází typicky k resorpci kostní tkáně osteoklasty, která vede ke vzniku osteolytických kostních ložisek. Osteoklasty jsou stimulovány k vyzrávání a aktivitě řadou cytokinů z nádorové tkáně, aktivita osteoblastů je utlumena. Protože se mnohočetný myelom vyskytuje převážně u starších jedinců, kdy je hematopoeticky aktivní dřeň již jen v axiálních částech kostry (páteř, sternum, žebra, lebka, lopaty kosti kýčelní), projevují se osteolytické kostní změny převážně v těchto částech kostry. Ty mohou způsobovat patologické fraktury, především kompresivní zlomeniny obratlů a zlomeniny žeber. Typickým klinickým příznakem MM jsou pak bolesti kostí, zejména páteře.
Mobilizace kalcia z kostí způsobuje hyperkalcémii. Té jsou přičítány některé psychické změny jako letargie, deprese, zmatenost. Hyperkalcémie se také spolupodílí na poškození renálních funkcí a na úporné zácpě.
Dalším typickým projevem MM je renální insuficience. Renální (tubulární) funkce mohou být poškozeny následkem selektivní proteinurie při vylučování lehkých imunoglobulinových řetězců lambda. Poškození tubulů se přičítá jednak jejich precipitaci v tubulech a jednak přetížení tubulárních buněk těmito paraproteiny. Poškození tubulů se nejčastěji projevuje tubulární acidózou, glykosurií, aminoacidurií a sníženou schopností koncentrovat moč.
Akutní myeloidní leukémie (AML) je heterogenní skupinou onemocnění (často definovaných specifickými genetickými změnami), které se jednotně vyznačují přítomností a akumulací atypických nezralých forem myeloidních buněk, patologických blastů, v kostní dřeni a většinou i v periferní krvi a event. ve slezině a v jiných orgánech (např. v játrech, meningách aj.). Jedná se o heterogenní skupinu onemocnění, u kterých vznikla patologická monoklonální hematopoeza v důsledku genetických změn (mutací), které postihly některou myeloidní krvetvornou buňku, ne nezbytně buňku kmenovou. Obecně se leukemický klon u AML vyznačuje nízkým až minimálním funkčním vyzráváním. Tento blok v diferenciaci spolu s proliferační výhodou vedou k akumulaci patologických blastů v kostní dřeni a útlaku normální krvetvorby.
!!!začátek petitu!!!
Ve většině případů procento myeloidních blastů v kostní dřeni přesahuje 20 % všech buněčných elementů, což je diagnostické pro AML. Méně často je procento blastů v kostní dřeni nižší než 20 %, ale je přítomna pro AML specifická genetická aberace, která vede k diagnóze AML. Někdy je možné určit, na základě určitého stupně diferenciace leukemických blastů, ze které vývojové řady leukemický klon vznikl. Jindy to možné není a pak můžeme předpokládat, že vznikl z málo diferencované buňky progenitorové nebo z buňky kmenové. Podle přítomnosti nebo chybění určitých diferenciačních znaků leukemických blastů je užívána morfologická klasifikace AML do osmi typů (tzv. FAB klasifikace, francouzsko-americko-britská).
!!!konec petitu!!!
Počet leukocytů v periferní krvi bývá většinou zvýšen, nejčastěji na hodnoty okolo 15 x 109/l krve, 25–40 % nemocných má však leukocytů v krvi méně než 5 x 109/l; v krvi jsou ovšem přítomny patologické leukemické blasty. Někteří nemocní pak mají více než 100 x 109 leukocytů/litr krve. Typickým laboratorním znakem je tzv. hiatus leukemicus, stav kdy je při vyšetření diferenciálního rozpočtu leukocytů v periferní krvi vedle zralých granulocytů různé množství atypických blastů, zatímco střední vývojová stadia granulocytů (tj. promyelocyty, myelocyty a metamyelocyty) chybějí.
Vedle morfologické klasifikace je dále užívána klasifikace, která dělí AML na dvě početné skupiny:
Pro ilustraci je uvedena v tabulce 1.5.
!!!tab. 1.5.!!!
Chromozomální abnormality (translokace, inverze, delece, monozomie, trizomie) jsou přítomny asi u 50 % případů AML (tab. 1.7). Pokud jsou přítomny 3 a více chromozomálních abnormalit, hovoříme o komplexních změnách karyotypu. Komplexní karyotyp bývá často doprovázen mutací genu TP53 a bývá asociován s velmi nepříznivou prognózou. U zbylých případů AML s normálním karyotypem bývá často detekována určitá mutace (např. mutace genu NPM1, CEBPA, KMT2A (MLL1), RUNX1, FLT3, RAS, WT1). Velmi zjednodušeně lze genetické alterace nacházené u AML dělit na dvě skupiny:
Přítomnost určitých genetických abnormalit u AML je významným prognostickým faktorem a má vliv na terapeutický přístup.
!!!začátek petitu!!!
Poměrně velmi dobře je známa molekulární podstata akutní promyelocytární leukémie (viz tab. 1.7). Gen PML je z 15. chromozomu přenesen na chromozom 17, kde se spojí s genem pro receptor z rodiny steroidních/tyreoidálních receptorů, označovaný jako RARα (retinoic acid receptor). Vznikne tak fúzní gen PML/RARα, jehož exprese je řízena promotorem genu PML. Normálně se RARα po navázání svého ligandu, kterým je forma vitaminu A – tzv. all-trans retinová kyselina (ATRA), váže ke specifickým nukleotidovým sekvencím v DNA a spolu s komplexem dalších proteinů umožňuje expresi genů důležitých pro diferenciaci a maturaci granulocytů. Vznikem fúzniho proteinu PML/RARα je fyziologická funkce RARα narušena, a porušena je proto i maturace myelocytů, které se hromadí ve stadiu odpovídajícím promyelocytu. Vysoké koncentrace ATRA obnovují funkci PML/RARα v granulocytové maturaci, a proto lze projevy promyelocytové leukémie touto látkou rychle a úspěšně léčit, tj. lze dosáhnout remise. Vlastní genetická porucha však touto léčbou odstraněna není. Na podávání vysokých dávek ATRA se může vyvinout rezistence, která se projeví relapsem této leukémie.
Existuje i forma promyelocytové leukémie, která je od počátku rezistentní k léčbě vysokými dávkami ATRA. V těchto případech je její příčinou jiná genetická změna, např. změna spojená s chromozomální translokací t(11,17). Při této translokaci se s genem RARα spojí gen z dlouhého raménka 11. chromosomu označovaný jako PLZF. Produkt fúzního genu, PLZF/RARα, rovněž naruší maturaci myelocytů. Produkt normálního genu PLZF je součástí transkripčních komplexů, které potlačují aktivitu (expresi) některých genů, jejichž produkty jsou významné pro buněčný cyklus. Produkt genu PLZF je tedy specifickým transkripčním inhibitorem a jeho fúze s genem RARα interferuje s touto jeho normální funkcí. Proliferace buněk příslušných k postiženému klonu se proto zvýší.
Při další chromozomální translokaci spojené se vznikem AML, translokaci t(8,21), vznikne fúzní gen AML1/ETO (RUNX1/RUNX1T), jehož produkt reaguje s transkripčním inhibitorem PLZF a znemožňuje jeho vazbu do regulačních komplexů jaderné matrix. Podobně může produkt fúzního genu AML1/ETO reagovat s produktem genu c/EBPα, který normálně působí jako diferenciační faktor, a to řízením exprese genů potřebných pro granulocytovou diferenciaci a maturaci. To je pravděpodobně další cesta, kterou translokace t(8,21) vyvolává AML. Podobně působí inaktivační mutace genu c/EBPα.
!!!konec petitu!!!
Hlavními klinickými projevy AML jsou příznaky způsobené potlačením erytropoezy s výslednou anémií, která bývá normochromní, normocytární s nízkým počtem retikulocytů, zvýšeným výskytem infekcí (zvláště bakteriální a plísňová onemocnění kůže, sliznic, plic) z důvodu nedostatku funkčních granulocytů, event. monocytů, a trombocytopenií (krvácivé projevy). Vysoký počet myeloidních blastů v periferní krvi může vést k příznakům souvisejícím s hyperviskozitou (dušnost, neurologické příznaky). Přítomnost blastů v periferní krvi může též způsobit diseminovanou intravaskulární koagulaci, což bývá časté u akutní promyelocytární leukémie. Dále může být přítomna hepatosplenomegalie, infiltrace měkkých tkání – např. zbytnění dásní, které odrážejí infiltraci těchto orgánů a tkání leukemickým klonem.
Vetšina AML vzniká de novo, bez předcházejícího onemocnění. Část případů AML se vyvjíjí z předchozího myelodysplastického syndromu nebo po léčbě jiného onemocnění, při níž dochází k expozici cytostatikům s alkylačními účinky, inhibitorům topoizomerázy II, ionizujícímu záření. Vliv může mít také expozice některým chemikáliím, z nichž nejznámější je benzen. Tato tzv. sekundární AML má většinou velmi nepříznivou prognózu.
Existují i případy vrozené predispozice ke vzniku AML. Jsou to např. syndrom familiární monosomie 7. chromosomu, trisomie 21. chromosomu (Downův syndrom), neurofibromatóza I. typu (Recklinghausenova nemoc), těžké vrozené neutropenie a Fanconiho anémie. Tyto predispozice jsou vysvětlovány vrozenou mutací jedné alely významného genu, u kterého obě alely musí být mutovány, aby vznikl patologický fenotyp – v tomto případě AML. Zatímco u zdravých homozygotů musí dojít k náhodné mutaci obou alel (pravděpodobnost je malá), u zdravých heterozygotů stačí jeden náhodný děj k tomu, aby se postižená buňka začala chovat jako leukemická kmenová buňka (pravděpodobnost, že toto nastane, je významně vyšší). Vznik leukemického klonu je pak u těchto geneticky predisponovaných jedinců výsledkem fenoménu označovaného jako ztráta heterozygotnosti.
Primární (idiopatická) myelofibróza (PM) je dalším myeloproliferativním onemocněním vznikajícím v důsledku genetických změn myeloidního progenitoru nebo hematopoetické kmenové buňky. Obdobně jako u ET, PM je asociována s přítomností určitých genetických změn: aktivační mutace genu JAK2 (V617F; asi u 50 % případů), mutace genu CALR (20-40 %) nebo aktivační mutace genu MPL pro trombopoetinový receptor (5-10 %).
Patogeneze tohoto onemocnění spočívá v postupném zániku krvetvorby v kostní dřeni s její současnou aktivací ve slezině, játrech a případně v dalších orgánech. Součástí patogeneze je tedy rozvoj extramedulární hematopoezy. Ta na rozdíl od normální hematopoezy umožní uvolnění malého počtu erytroblastů, myelocytů a promyelocytů do cirkulace, které normálně v periferní krvi nenalézáme. Erytrocyty mohou být charakteristicky deformované do podoby kapky (angl. teardrops). V kostní dřeni se hromadí vazivová tkáň bohatá na kolagen III. Kolagen je produkován reaktivními fibroblasty. Fibroblasty jsou stimulovány růstovými faktory, které jsou sektretovány abnormálními megakaryocyty. Jedná se zejména o růstový faktor PDGF, bazický fibroblastový růstový faktor (bFGF) a cytokin TGF-β.
Rozvinuté onemocnění se projevuje zejm. anémií, splenomegalií a hepatomegalií (může být přítomna portální hypertenze s ascitem), hubnutím, horečkami, nočním pocením. Může docházet k trombotickým či krvácivým projevům, častějším infekcím. Asi jedna třetina nemocných progreduje do akutní myeloidní leukémie.
Mezi myeloproliferativními onemocněními má PM nejhorší prognózu a u části nemocných s vysokým rizikem je indikována alogenní transplantace krvetvorných buněk.
Lymfoproliferativní onemocnění jsou nádorová onemocnění, která vznikají maligní transformací různých vývojových stádií lymfocytů. Velmi zjednodušeně tak lze z patofyziologického hlediska lymfoproliferativní onemocnění dělit na:
A. Prekurzorové a periferní lymfoproliferace (podle toho zda vznikají z nezralého lymfoidního prekurzoru či periferních/zralých lymfocytů)
B. B- a T-lymfoproliferace (podle toho, zda onemocnění vzniká z B či T lymfocytární vývojové řady).
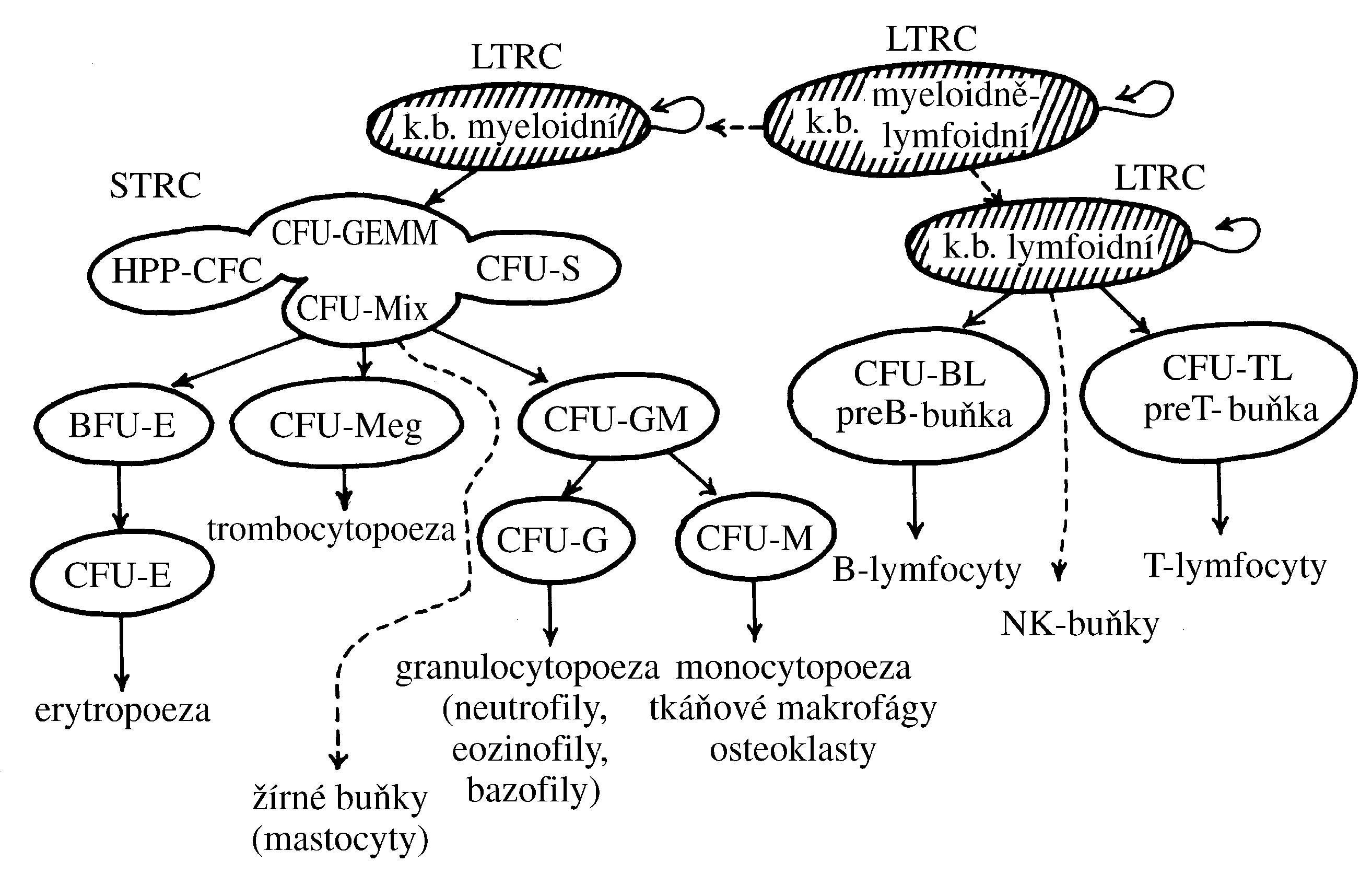
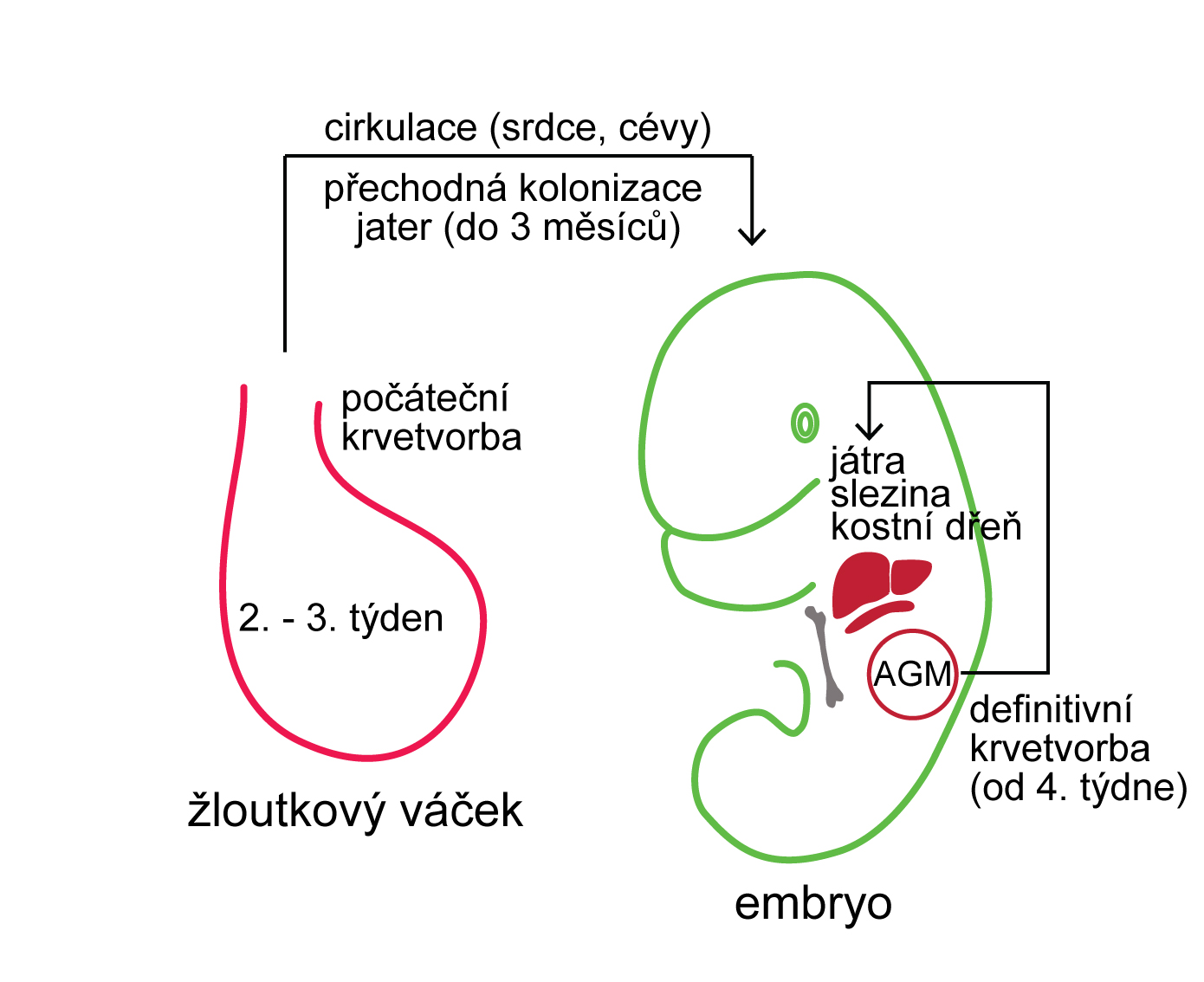
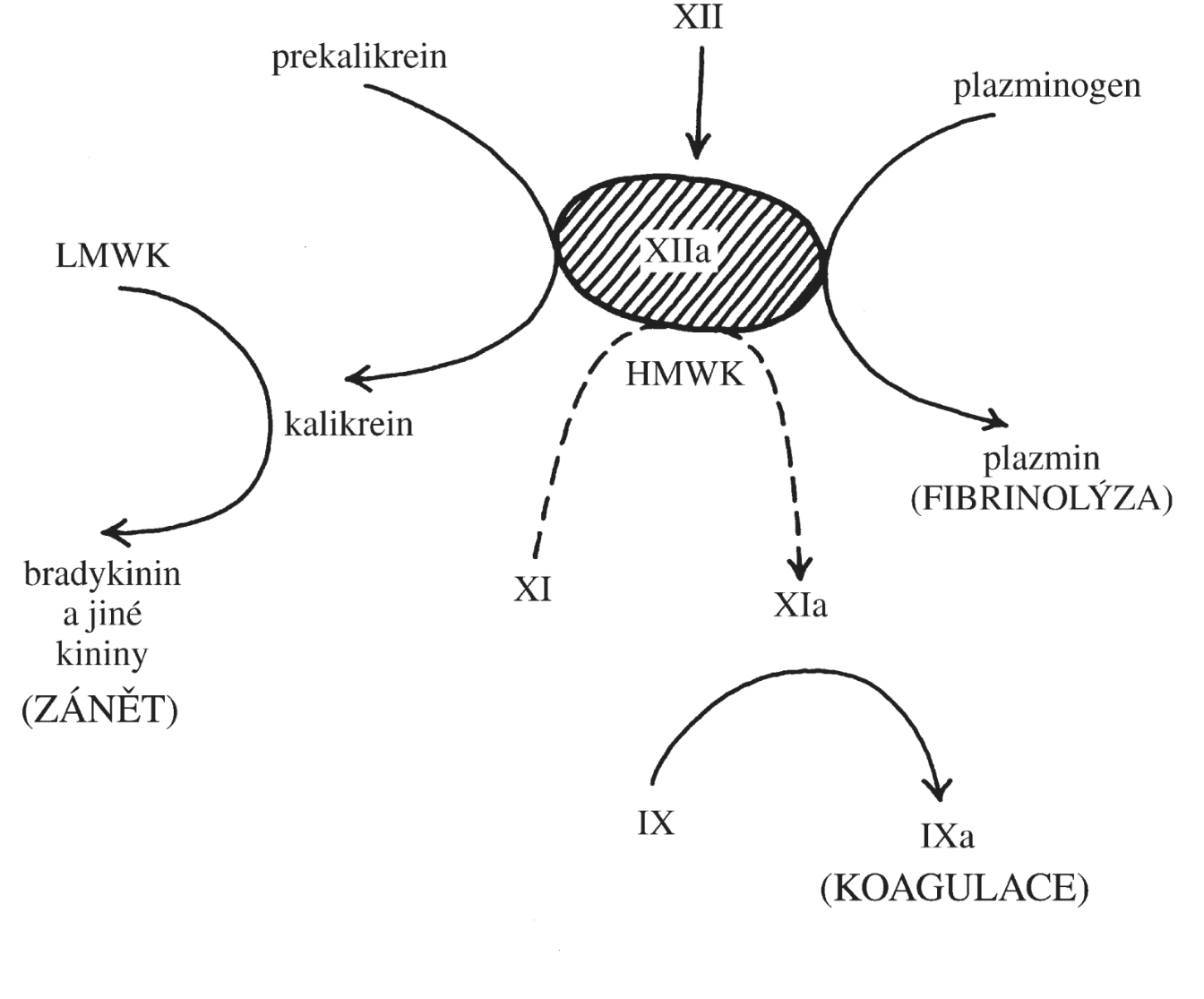
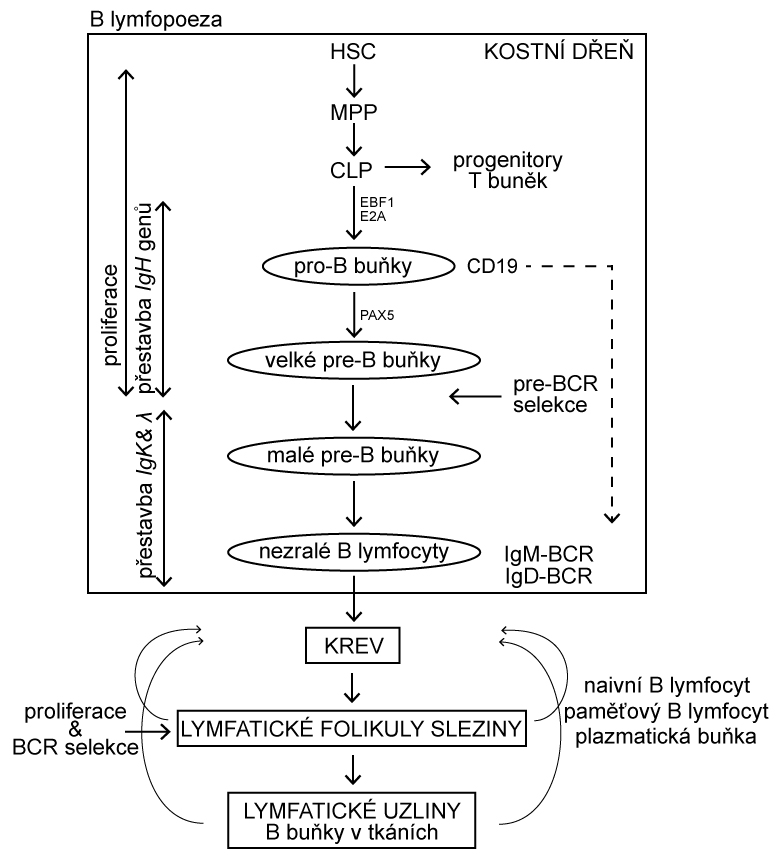
Pro pochopení patogeneze lymfoproliferativních onemocnění je nutná znalost normálního vývoje B- a T-lymfocytů. B a T lymfocyty vznikají několikastupňovým vývojem ze společeného lymfoidního prekurzoru, který vzniká v kostní dřeni diferenciací z multipotentní hematopoetické kmenové buňky (Obr. 1.9). Pro vývoj B lymfocytů je zásadní vyzrávání B buněčného receptoru (BCR, z angl. B cell receptor). BCR je nezbytný pro vývoj i imunitní funkce B lymfocytů a je tvořen dvěma těžkými (IGH) a dvěma lehkými (IGL) řetězci. Těžký a lehký řetězec BCR je kódován odlišnými geny (lokus IGH pro těžký řetězec; IGK nebo IGL pro lehký řetězec). Specifickou vlastností B lymfocytů je, že geny pro oba řetězce BCR jsou modifikovány specializovaným enzymatickým aparátem, který odstraňuje a opětovně spojuje jednotlivé V (variable), D (diversity) a J (joining) části genu v případě těžkého řetězce nebo V a J části v případě řetězce lehkého a vytváří tak variabilní část BCR. Proces V(D)J rekombinace zajišťuje dostatečný repertoár B buněčných receptorů pro reakci s různými antigeny a umožňuje tak účinně a specificky reagovat např. na přítomnost patogenu v organismu. V(D)J rekombinace je iniciována působením komplexu enzymů zahrnujících proteiny RAG1 (recombination activating gene) a RAG2 a dochází při ní ke dvouvláknovým zlomům DNA a následné opravě DNA mechanismem nehomologního spojování konců DNA. Právě tyto modifikace DNA však mohou predisponovat ke vzniku genetických změn souvisejících s maligní transformací vyvíjejícího se lymfocytu. Chybně probíhající VDJ rekombinace tak může být jednou z příčin vzniku lymfoproliferativních onemocnění (lymfatické leukémii a lymfomů).
!!!obr. 1.9.!!!
Pouze ty B lymfocyty, které provedly V(D)J rekombinaci úspěšně tzn. je zachován čtecí rámec genů obou podjednotek receptoru a produkuje se funkční protein, mohou opustit kostní dřeň a následně dozrávat v sekundárních lymfoidních orgánech, slezině a lymfatických uzlinách.. Tyto tzv. naivní B lymfocyty se v sekundárních lymfoidních orgánech setkávají s antigenem, specifickým z hlediska variabilní části jejich BCR, který je stimuluje k aktivitě a a proliferaci, při kterých dochází k řízenému zásahu do jejich genetické informace tzv. somatickou hypermutací (SHM, z angl. somatic hypermutation) a izotypovým přesmykem (CSR, z angl. class switch recombination). Proces SHM je zprostředkován enzymem AID a dochází při něm k mutacím (bodové mutace, delece, inzerce) v genech kódujících variabilní část lehkého a těžkého řetězce BCR. To zajišťuje produkci protilátek s vysokou afinitou vůči imunizujícímu antigenu. SHM však může proběhnout chybně a může tak docházet k bodovým mutacím řady genů, včetně některých protoonkogenů. Izotypový přesmyk je také zajišťován enzymem AID a dochází při něm k přestavbě genových segmentů pro konstantní části těžkého řetězce imunoglobulinu (variabilní doména je zachována). Izotypový přesmyk umožňuje náhradu původní konstantní části imunoglobulinu (IgM, IgD) za jiný izotyp (IgA, IgE, IgD) a tím získat produkovanému imunoglobulinu další vlastnosti, např. v případě izotypu IgA schopnost přecházet na slizniční povrchy. Bylo prokázáno, že abnormálně probíhající izotypový přesmyk může způsobit některé chromozomální translokace. Z popsaného vyplývá, že při vývoji B lymfocytů dochází opakovaně k zásahům do genetické informace již zmíněnými mechanismy V(D)J rekombinace, SHM a CSR, které mohou způsobit chromozomální translokace či mutací protoonkogenů a vzniku lymfoproliferativních onemocnění. Vznik nádorového onemocnění je však vícestupňový proces a pro maligní transformaci vyvíjejícího se lymfocytu je většinou nutná akumulace vyššího počtu onkogenních genetických změn. Řada genetických změn také vyvolá apoptózu postižené buňky, která zamezí vzniku patologického klonu buněk.
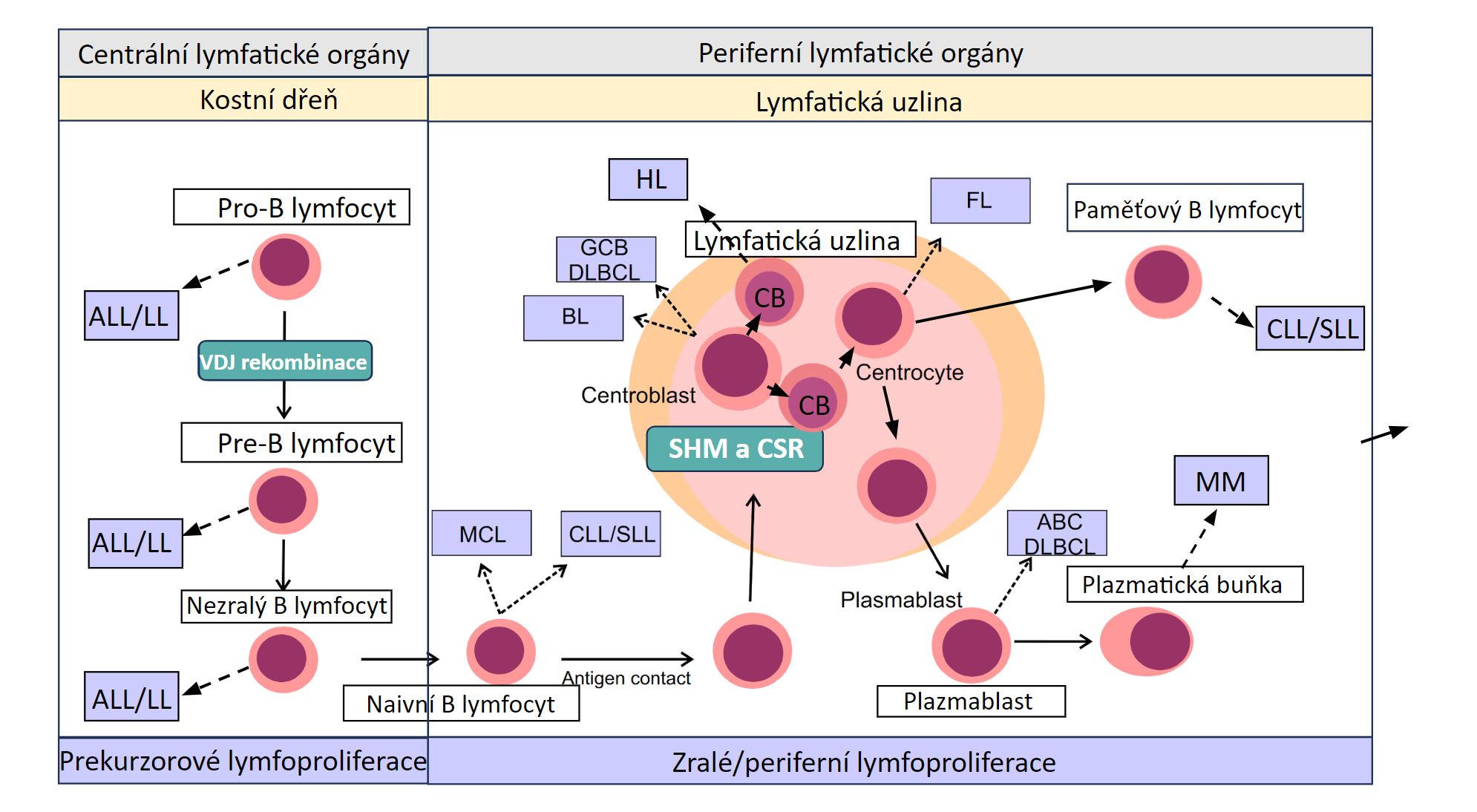
K maligní transformaci vyvíjejícího se lymfocytu může dojít na jakémkoliv stupni jeho vývoje, řada lymfoproliferací má tedy své fyziologické protějšky z hlediska stupně vyzrávání B lymfocytu. Lymfoproliferace odvozené od určitého vývojového stupně B lymfocytu si zachovávají některé jeho charakteristiky – expresi povrchových receptorů, genovou expresi, charakter růstu, atd. Tyto charakteristiky jsou důležité pro diagnostiku a klasifikaci jednotlivých podtypů B lymfoproliferací. Na obrázku 1.10 jsou jednotlivá vývojová stadia B lymfocytů a z nich odvozená lymfoproliferativní onemocnění.
!!!obr. 1.10.!!!
!!!začátek petitu!!!
Vzhledem k tomu, že při normálním vývoji B lymfocytů dochází opakovaně k řízeným zásahům do DNA kódující imunoglobulinové geny, není překvapivé, že u B lymfoproliferací jsou velmi časté chromozomální translokace, které zahrnují imunoglobulinové geny. Při těchto genetických změnách dochází k přenesení některého buněčného protoonkogenu (např. genu BCL2, CCND1, MYC) do oblasti genomu, která kóduje některý imunoglobulinový řetězec. Jedná se např. o t(14;18), t(11;14) nebo t(8;14), viz dále.
!!!konec petitu!!!
V tabulce 1.6 jsou vybraná lymfoproliferativní onemocnění, nejčastější genetické změny – chromozomální translokace a jiné genetické alterace a další základní charakteristiky.
!!!Tab. 1.6.!!!
Na rozdíl od B lymfocytů, progenitory T lymfocytů, které vznikají ze společného lymfoidního prekurzoru v kostní dřeni, opouštějí kostní dřeň a dále se vyvíjí v thymu. Podobně jako při vývoji B lymfocytů i T lymfocyty potřebují na svém povrchu funkční T buněčný receptor pro vazbu antigenu. Velká diverzita T buněčných receptorů pro reakci s různorodými antigeny je také zajišťována procesem VDJ rekombinace 4 genů pro alfa, beta, gama a delta řetězce. Zralé T lymfocyty pak exprimují buď T buněčný receptor složený z podjednotek αβ nebo receptor složený z podjednotek γδ. Na rozdíl od B lymfoproliferací, u nichž je možná klasifikace na základě vývojového stupně B lymfocytu, od něhož je daná malignita odvozena, toto je u T lymfoproliferací mnohem obtížnější vzhledem k velké diverzitě T lymfocytů a dalším faktorům, které se při maligní transformaci uplatňují.
!!!začátek petitu!!!
U Burkittova lymfomu a v některých případech akutní lymfoblastové leukémie a některých případech difúzního velkobuněčného B lymfomu je v buňkách patologického klonu přítomna translokace t(8;14), t(2;8) nebo t(8;22). Tyto translokace způsobují, že buněčný protoonkogen c-myc je přenesen z chromosomu 8 do oblasti chromosomu 14 kódující těžký imunoglobulinový řetězec nebo do oblastí chromosomů 2 a 22, které kódují lehké imunoglobulinové řetězce κ a λ. c-MYC je transkripční faktor, který reguluje expesi mnoha genů, které zajišťují buněčnou proliferaci, růst nebo regulují apoptózu.
Při chromosomální translokaci t(14;18), přítomné u většiny případů folikulárního lymfomu a asi u 25 % případů difúzního velkobuněčného B-lymfomu, je protoonkogen bcl-2 přenesen z chromozomu 18 do oblasti kódující těžký imunoglobulinový řetězec na chromozomu 14. Nadměrná exprese genu bcl-2 zvyšuje odolnost postižených buněk k apoptóze, což v lymfatické tkáni, kde existuje významná fyziologická apoptóza, způsobuje hromadění maligních lymfocytů. Zvýšená rezistence vůči apoptóze však také snižuje pravděpodobnost eliminace buňky, ve které vznikne další genetická porucha. V populaci (klonu) lymfoidních buněk se zvýšenou expresí bcl-2 je proto zvýšená nestabilita genomu.
Pro lymfom z plášťových buněk je charakteristická chromozomální translokace t(11;14), při níž dochází k fúzi genu, který kóduje cyklin D1 s regulační sekvencí genu pro těžký imunoglobulinový řetězec. Tato chromozomální aberace vede ke konstitutivně zvýšené expresi cyklinu D1 a stimulaci buněčného cyklu z fáze G1 do S, což se projeví zvýšením proliferace maligních lymfocytů.
!!!konec petitu!!!
Lymfoproliferativní onemocnění lze ještě dělit z klinického hlediska na agresivní a indolentní onemocnění. Agresivní lymfoproliferace (např. akutní lymfoblastová leukémie, lymfoblastový lymfom, difúzní velkobuněčný lymfom) se vyznačují rychlým nástupem klinických příznaků a nutností neprodleně zahájit léčbu tohoto onemocnění. Naopak indolentní lymfoproliferace (např. chronická lymfocytární leukémie, folikulární lymfom) mají nástup klinických příznaků pozvolný, často mohou být diagnostikovány náhodně při podezření na jiné onemocnění a v léčebné strategii lze někdy aplikovat přístup „watch and wait“ – tj. sledování onemocnění s odkladem terapie na dobu další progrese onemocnění. Indolentní lymfoproliferace (např. chronická lymfocytární leukémie, folikulární lymfom) mohou transformovat do agresivní lymfoproliferace (nejčastěji do difúzního velkobuněčného B-lymfomu). Tento proces je způsoben akumulací dalších genetických změn, které změní vlastnosti nádorového klonu (např. zvýšení proliferace nádorových buněk).
Jsou známy některé vlivy, které riziko vzniku lymfoproliferativních onemocnění zvyšují. Jsou to dlouhodobá imunosupresivní léčba, infekce HIV (zejména u agresivních B-lymfomů), celotělové ozáření, předchozí léčba cytostatiky, autoimunitní onemocnění, virová infekce EBV (u Burkittova lymfomu, zvláště afrického typu), infekce virem HTVL-1 (v případě T-buněčných akutních lymfoblastických leukémií vyskytujících se v části Japonska, Karibiku a USA), a další. Genetická predispozice je málo častá, typicky se vyskytuje v menšině případů chronické lymfocytární leukémie.
9Není to zcela specifický projev této skupiny nemocí. Může se vyskytnout i u CLL, lymfomů, ale i u nenádorových onemocnění: jaterní cirhózy, sarkoidózy aj.
Při těchto poruchách dochází k hromadění některých buněk příslušejících k myeloidní vývojové řadě. Tyto buňky jsou většinou více nebo méně odlišné od buněk normálních. Od normálních buněk se často liší vyšší proliferační aktivitou, vyšší odolností k apoptóze, nižší adhezivitou ke stromatu krvetvorné tkáně a nedokonalým funkčním vyzráváním4.
10Jedná se o relativní jednotky, které porovnávají viskozitu plazmy s viskozitou destilované vody (ta je 1,0).
Waldenstromova makroglobulinémie (WM) vzniká maligní transformací vyvíjejících se B lymfocytů, které prošly zárodečným centrem sekundárních lymfoidních orgánů, prodělaly somatickou hypermutaci, nikoliv však izotypový přesmyk. Pro toto onemocnění je typická přítomnost mutace genu MYD88 (kóduje stejnojmenný protein).
!!!začátek petitu!!!
MYD88 je protein, který po vazbě ligandu přenáší signál z intracelulární strany tzv. toll-like receptorů (TLR; slouží k vazbě antigenu a jsou exprimovány zejména buňkami imunitního systému) dále do buňky a zahajuje aktivaci signální dráhy NFκB. Mutace genu MYD88 nacházené u WM způsobují aktivaci NFκB dráhy nezávisle na stimulaci TLR.
!!!konec petitu!!!
Patologický klon produkuje paraprotein typu IgM. Na rozdíl od mnohočetného myelomu je patologický klon lymfoplazmocytů nejen v kostní dřeni, ale i ve slezině, v játrech, v lymfatických uzlinách a také v krvi. Hepato/splenomegalie a lymfadenopatie patří mezi symptomy tohoto onemocnění. Další odlišnost od mnohočetného myelomu spočívá v tom, že nebývá narušen kostní metabolismus. M-komponenta je tvořena velkou molekulou IgM, a proto nebývá proteinurie, nebo je malá. Množství paraproteinu v plazmě často převyšuje 30 g/l a mohou tedy být známky hyperviskozity. Část paraproteinu má charakter kryoglobulinu. U nemocných se proto může projevit Raynaudův fenomén. Častá je i anémie a zvýšená krvácivost (hemoragická diatéza).
Obrázek 10 Podrovnosti viz text
4Nejednou se jedná o skupinu onemocnění heterogenních svou specifickou genetickou změnou i fenotypem.
Systémová mastocytóza (SM) je vzácné onemocnění, agresivní forma SM má velmi špatnou prognózu. SMje charakterizována proliferací a akumulací žírných buněk (mastocytů), a to jednak v kostní dřeni, jednak v různých tkáních a orgánech, nejčastěji slezině, játrech, lymfatických uzlinách a sliznici gastrointestinálního traktu. Mastocyty mají membránový receptor c-Kit, což je receptor pro růstový faktor SCF. V etiologii a patogenezi systémové mastocytózy se ve většině případů uplatňuje aktivační bodová mutace genu pro c-Kit. Ta působí na ligandu nezávislou aktivaci c-Kit s výslednou proliferací a akumulací žírných buněk.
Klinické projevy mastocytózy zahrnují symptomy způsobené látkami uvolňovanými z mastocytů při jejich degranulaci. Mezi tyto látky patří i histamin. Tyto symptomy zahrnují: záchvaty svědění, záchvatovité zčervenání kůže, záchvaty arteriální hypotenze, křečovité bolesti břicha, průjmy s malabsorbcí v gastrointestinálním traktu, bolesti hlavy. Žaludeční hypersekrece (stimulace sekrece HCl histaminem), gastritida případně vředová choroba žaludku jsou proto dalším logickými projevy tohoto onemocnění. Systémová mastocytóza může vyvolat fibrotické změny v játrech, ve slezině a v kostní dřeni. Hromadění mastocytů v kůži se projevuje jako červeno-hnědé makuly a papuly (urticaria pigmentosa). Dalšími nespecifickými příznaky je hubnutí a noční pocení.
Primární amyloidóza je onemocnění, u kterého dochází k depozici nerozpustných fibril amyloidu v různých tkáních a orgánech, což vede k jejich dysfunkci a později orgánovému selhání. U primární amyloidózy jsou fibrily amyloidu tvořeny monoklonálními lehkými imunoglobulinovými řetězci (obvykle λ, méně často κ). Zdrojem patologických imunoglobulinových řetězců je patologický klon plazmatických buněk. Primární amyloidóza může doprovázet různá lymfoproliferativní onemocnění, ale většinou vzniká z monoklonální gamapatie nejistého významu (MGUS).
Symptomy tohoto onemocnění se odvíjí od postižení a dysfunkce jednotlivých orgánů a tkání. Nejčastěji dochází k postižení ledvin (projevuje se např. jako nefrotický syndrom, renálním selháním), srdce (vede k arytmiím, srdečnímu selhání), jater (hepatomegalie, laboratorní zvýšení jaterních enzymů), gastrointestinálního traktu (projevuje se např. malabsorpcí, gastroparézou, zácpou), infiltrací měkkých tkání (makroglosie).
Tabulka 1.6 Chromozomální translokace u vybraných B-NHL
| Lymfoproliferace | Chromozomální translokace | Deregulovaný onkogen | Funkce | Jiné genetické alterace | Klinické chování |
| DLBCL | t(14;18)# | BCL2 | Inhibice apoptózy | KMT2D mut, CDKN2A del*, TP53 mut/del, MYD88 mut* | agresivní |
| t(3;14)* | BCL6 | TF, tolerance k poškození DNA | |||
| t(8;14), t(2;8),t(8;22) | MYC | TF, Proliferace | |||
| FL | t(14;18) | BCL2 | Inhibice apoptózy | KMT2D mut, CREBBP mut, TNFRSF14 mut, EZH2 mut | indolentní |
| MCL | t(11;14) | Cyklin D1 | Stimulace buněčného cyklu | TP53 mut/del, CDKN2A/2B del, ATM mut/del | agresivní |
| BL | t(8;14), t(2;8),t(8;22) | MYC | TF, Proliferace | agresivní | |
| CLL | - | 13q14 del (mIR15, mIR16), TP53 mut/del, ATM del/mut | indolentní | ||
| HL | - | 9p24.1 amplifikace (PDL1, JAK2) | agresivní |
BL – Burkittův lymfom; CLL – chronická lymfocytární leukémie; DLBCL – difúzní velkobuněčný lymfom; FL – folikulární lymfom; HL – Hodgkinův lymfom, MCL – lymfom z plášťových buněk; TF – transkripční faktor; # významně častější u GCB DLBCL ve srovnání s ABC DLBCL, *významně častější u ABC DLBCL ve srovnání s GCB DLBCL.
T
66letý muž byl vyšetřen pro zhoršující se únavnost, slabost a recidivující zvýšené teploty. Byla zjištěna leukopenie. Podrobnější hematologické vyšetření, které zahrnulo i sternální punkci a vyšetření vzorku kostní dřeně, vedlo k diagnóze myelodysplastického syndromu s klasifikací IB-1. Po léčbě azacitidinem (antimetabolit, analog cytidinu) bylo dosaženo remise. Po několika měsících byl pacient hospitalizován z důvodu horečnatého onemocnění s teplotami až 39,2 °C.
Výsledky fyzikálního a laboratorního vyšetření:
Krevní obraz (tab. K1.1)
Protilátky proti neutrofilním granulocytům ani trombocytům nebyly prokázány. Přímý Coombsův test neprokázal přítomnost protilátek na erytrocytech. Nepřímý Coombsův test byl slabě pozitivní.
Pacient byl léčen antibiotiky a chemoterapií zaměřenou na základní onemocnění. Dostal opakované transfúze erytromasy a trombocytových náplavů. V moči byla prokázána přítomnost bakterie Staphylococcus aureus, hemokultura byla pozitivní na Staphylococcus haemolyticus. Po léčbě antibiotiky se staly jak moč, tak hemokultura přechodně na přítomnost bakterií negativní. Dostavily se však znovu vysoké teploty a hemokultura byla pozitivní na Corynebacterium, později na Candida albicans. Desátý den hospitalizace se dostavila protrahovaná epistaxe. Později nastaly poruchy vědomí a 42. den hospitalizace pacient zemřel.
Patofyziologický rozbor
Porucha maturace myeloidních prekurzorů, typická pro myelodysplastický syndrom, způsobila anémii, leukopenii a trombocytopenii. Počáteční potíže, zhoršující se únavnost, slabost a recidivující teploty, lze přičítat těmto změnám krevního obrazu.
Vedle anémie, leukopenie a trombocytopenie jsou v krevním obrazu velmi nízké hodnoty výskytu retikulocytů, zvláště přihlédneme-li k asi polovičnímu počtu erytrocytů. Očekávaný výskyt retikulocytů při tomto stupni anémie a při normální produkci erytrocytů (retikulocytů) by byl asi 2–3 %. Hlavním patogenetickým mechanismem anémie byla proto snížená produkce erytrocytů krvetvornou tkání. Hodnoty červeného krevního obrazu byly samozřejmě ovlivněny opakovanými transfúzemi erytromasy. Bez nich by nepochybně byla anémie ještě výraznější. Střední objem erytrocytů (MCV) byl lehce zvětšen, jak je pro MDS typické.
Leukopenie byla zřetelná, ale většinou nebyla pod udávanou kritickou hodnotou 0,5 x 109/l krve6. Přesto měl pacient opakované infekce, které byly provázeny bakteriémií a zvýšenými teplotami, které byly charakterizované jako horečky septického charakteru. Vysvětlení je třeba hledat ve velmi nízkém zastoupení granulocytů mezi leukocyty krve. Jejich normální zastoupení v diferenciálu bílých krvinek krve je 60–70 %, u pacienta byly zastoupeny jen asi 10 nebo méně procenty. Převažujícím typem leukocytů byly lymfocyty (jejich zastoupení je normálně 20–25 %). Odhad absolutního počtu granulocytů v krvi, učiněný z celkového počtu leukocytů a procentuálního zastoupení granulocytů, je 0,24–0,06–0,02 x 109/l, při kritické hodnotě asi 0,5 x 109/l a normální hodnotě asi 3,5 x 109/l. Granulocytopenie byla tedy velmi výrazná, což vysvětluje náchylnost k infekcím. Praktické chybění tyček v diferenciálu ukazuje na velmi nízkou produkci granulocytů.
Trombocytopenie byla výrazná přes transfúze trombocytů. Projevem hemoragické diatézy bylo protrahované krvácení z nosu, epistaxe.
Poruchy vědomí a smrt pacienta pravděpodobně způsobovaly infekce s opakovanými septickými epizodami. Nelze vyloučit ani možnost krvácení do CNS. Akutní ohrožení krvácení do mozku nastává při počtu trombocytů < 10 x 109/l krve.
Komentář
Klinické projevy myelodyplastického syndrom, jakož i pancytopenie myeloidních buněk (erytrocytů, granulocytů a trombocytů) a nízký počet retikulocytů a tyček v krvi, jsou podobné změny, jaké se vyskytují při aplastické anémii (viz kap. 1.4.4). Kostní dřeň však není při myelodysplastickém syndromu chudá na jaderné prekurzory krevních buněk a jsou v ní přítomny patologické, blastům podobné, buňky. Při jednom vyšetření diferenciálu leukocytů byly prokázány blasty i v periferní krvi.
Myelodysplastický syndrom (MDS) je heterogenní skupinou hematologických malignit, které jsou způsobeny získanými genetickými změnami (mutacemi), které postihují hematopoetickou kmenovou buňku. Tyto mutace vedou k poruše normálního vyzrávání (diferenciace), dysplázii (morfologické abnormality) a zvýšené proliferaci buněk patologického klonu. Pro MDS je typická neefektivní hematopoéza – stav, kdy buňky příslušející k patologickému klonu zvýšeně podléhají apoptóze a tudíž, přes zvýšenou proliferaci, není produkce zralých krevních buněk dostatečná. MDS bývá proto v krvi asociován s anémií (často makrocytární), leukopénií, trombocytopenií či pancytopenií. Kostní dřeň bývá přitom normocelulární, nebo dokonce vzhledem k věku hypercelulární5. MDS je asociován s vysokým rizikem transformace do akutní myeloidní leukémie (AML).
Patologická hematopoeza v kostní dřeni je typicky monoklonální. Vyšší věk je nejdůležitějším rizikovým faktorem pro vznik MDS. To je připisováno progresivní akumulaci somatických mutací hematopoetických kmenových buněk, ke které dochází v průběhu života.
!!!začátek petitu!!!
Mutace či kombinace mutací může dát vzniknout buněčnému klonu, který má odolnost vůči apoptóze a proliferační výhodu. Vzniká tak klonální hematopoéza (detekovatelná u > 10% populace nad 70 let). Tento abnormální buněčný klon je pak v riziku akumulace dalších mutací, které zvyšují jeho maligní potenciál a mohou způsobit některou formu MDS.
!!!konec petitu!!!
U MDS často nacházíme chromozomální aberace (např. delece 5q, delece 7q, trizomie chromozomu 8) nebo bodové mutace genů, které regulují methylaci DNA (např. TET2, DNMT3A), sestřih RNA (SF3B1) či modifikace chromatinu (ASXL1). Přítomnost či nepřítomnost genetických změn je důležitá k určení prognózy pacientů s MDS.
Ve většině případů vzniká MDS sporadicky. Některé případy MDS však vznikají po předchozí expozici cytostatikům s alkylačním účinkem, ionizujícímu záření, benzenu aj. Dalšími faktory, které zvyšují výskyt MDS, jsou některé vrozené genetické poruchy (trizomie 21. chromosomu – Downův syndrom, Fanconiho anémie, neurofibromatóza 1. typu – Recklinghausenova nemoc).
Klasifikace MDS je velmi komplexní a vychází zejména z morfologických (např. % nezralých buněk, blastů, v kostní dřeni či periferní krvi), cytogenetických nálezů (přítomnost chromozomálních aberací) a molekulárně-biologických nálezů (detekce specifických mutací).
Ke klinickým příznakům patří zejména příznaky spojené s anémií (únava, dušnost, bledost kůže a sliznic), trombocytopenií (krvácivé projevy) a leukopenií (časté infekce). Asi v jedné třetině případů přechází MDS v některou formu akutní myeloidní leukémie. AML vzniklá transformací z MDS bývá prognosticky velmi nepříznivá.
6Ta samozřejmě platí pro normální diferenciál leukocytů, jak je dále rozvedeno.
Jedná se o orientační hodnocení nátěrů kostní dřeně na základě zkušenosti, aniž by byl počet jaderných buněk přesněji kvantifikován. U asi 10 % nemocných s MDS má však kostní dřeň charakter hypocelulární.
Tab. K1.2 Hematologické vyšetření
| Den (vztažený k úrazu a operaci) | |||||
| 6 měsíců | 1. | 5. | 9. | 13. | |
| Hemoglobin (g/l) | 142 | 104 | 68 | 59 | 72 |
| Erytrocyty (x 1012/l) | 4,6 | 3,0 | 2,0 | 1,7 | 2,2 |
| Hematokrit (%) | 39 | 29 | 19 | 18 | 22 |
| MCV (fl) | 84 | 96 | 91 | 103 | 100 |
| MCH (pg) | 31 | 34 | 32 | 35 | 33 |
| Retikulocyty (%) | 0,9 | 2,1 | 4,0 | ||
| Leukocyty (x 109/l) | 3,5 | 2,3 | 1,6 | 2,3 | 3,8 |
| Neutrofilní granulocyty (%) | 76 | ||||
| Tyčky (%) | 3 | ||||
| Eozinofilní granulocyty (%) | 1 | ||||
| Bazofilní granulocyty (%) | 0 | ||||
| Monocyty (%) | 11 | ||||
| Lymfocyty (%) | 9 | ||||
| Trombocyty (x 109 /l) | 150 | 114 | 34 | 34 | 47 |
K1.2 Přechodná idiopatická aplastická anémie
21letý muž byl ošetřován pro poúrazovou tříštivou zlomeninu humeru. Během chirurgického výkonu bylo nápadné difúzní krvácení z měkkých tkání. Vyšetření krevního obrazu, provedené v souvislostí s operací, ukázalo nízké hodnoty v červeném krevním obrazu, leukocytů a trombocytů. Během operace byly podány transfúze erymasy. Následující den se objevil ikterus. Pátý den po operaci byl pacient unavený, docházelo k občasnému krvácení z nosu, měl večerní subfebrilie. Krevní obraz byl vyšetřen před půl rokem při darování krve a byl normální. Asi 14 dnů před úrazem prodělal infekční (virové?) onemocnění, léčené Paralenem (paracetamol) a vitaminy.
Výsledky vyšetření 5. den po operaci:
Krevní obraz (tab. K1.2)
Protilátky proti neutrofilním granulocytům ani trombocytům nebyly prokázány. Přímý Coombsův test neprokázal přítomnost protilátek na erytrocytech. Osmotická rezistence erytrocytů byla normální.
Koncentrace železa v plazmě byla 4,2 μmol/l (norma 8–26), transferinu 4,2 g/l (norma 1–3), sérového feritinu 673 ng/ml (norma 20–220), vitaminu B12 151 ng/l (norma 380–1500).
Výsledky koagulačních vyšetření byly normální: Quickův test INR 1,0, aPTT 27 s, trombinový čas 15,7 s, antitrombin III 91 % normálu, fibrinogen 4,7 g/l, D-dimer > 2000 μg/l.
Vyšetření vzorku kostní dřeně získané aspirační punkcí: nátěry byly chudé na buňky, erytroblasty byly více zastoupeny (normálně jsou zastoupeny 20 % všech jaderných buněk – viz obr. 1.3). Nebyly nalezeny patologické formy prekurzorů krevních buněk.
Protilátky proti viru hepatitidy A, B a C byly negativní, stejně jako antigen viru B (HBsAg). Negativní byly i protilátky proti cytomegaloviru, herpes simplex viru a viru Epsteina-Barrové.
Patofyziologický rozbor
Únava, subfebrilie, časté epistaxe a jiné krvácivé projevy mohou být způsobeny poruchami buněčné složky krve. Nález pancytopenie je třeba doplnit vyšetřením kostní dřeně, pro možnost aleukemické formy akutní myeloblastové leukémie, vyloučení myelodysplastického syndromu, u starších jedinců mnohočetného myelomu či jiné nádorové infiltrace kostní dřeně. Hypoplastická dřeň, ve které jsou přítomny nečetné, ale fenotypem normální prekurzory krevních buněk, svědčí pro aplastickou anémii jako příčinu pancytopenie.
Aplastická anémie může mít dlouhodobý charakter a může být smrtelným onemocněním pro příliš nízkou produkci krevních buněk. Může být také přechodná, jak tomu bylo v tomto případě. Přítomnost retikulocytů v krvi ukazovala na významnou produkci erytrocytů, která 13. den dosahovala přibližně normálních nebo lehce zvýšených hodnot (4 % retikulocytů by byly asi 2 % při normálním počtu erytrocytů, tedy normální nebo lehce zvýšené hodnota). Především tento nález svědčí z dostupných výsledků na přechodný charakter onemocnění.
Výsledky vyšetření nesvědčily pro účast zvýšených ztrát erytrocytů na vzniku anémie (test na okultní krvácení, absence protilátek proti krevním buňkám, normální osmotická rezistence erytrocytů). Po operaci se však zvýšila hladina celkového bilirubinu v plazmě a objevil se ikterus. To bylo přičítáno hemolýze části erytrocytů podaných ve formě transfúze erytromasy (4 jednotky) během operace. Při nekomplikované transfúzi by se však koncentrace bilirubinu tak výrazně zvýšit neměla.
Pokles koncentrace hemoglobinu mezi 1. a 5. dnem (ze 104 g/l krve na 68 g/l krve) je příliš velký na to, aby ho bylo možné přičíst jen snížení tvorby erytrocytů. Je proto třeba předpokládat, že došlo ke zvýšeným ztrátám erytrocytů následkem jejich hemolýzy nebo následkem krvácení.
Laboratorní vyšetření ukázala nízkou hodnotu hladiny sérového železa, zvýšenou hladinu transferinu a zvýšenou koncentraci sérového feritinu. Tento nález jen částečně odpovídá redistribuci zásobního železa v organismu, ke kterému dochází při zánětových onemocněních. Železo je pevněji vázáno v zásobní formě feritinu, při současném snížení saturace transferinu železem a snížené koncentraci železa v plazmě. Zvýšená hladina transferinu v plazmě může být reakcí na relativní nedostatek železa. Transferin je však negativní protein akutní fáze, proto bychom v pooperačním období očekávali spíše snížení jeho hladiny. Zvýšení koncentrace C-reaktivního proteinu odpovídá pooperačním stavu, protože se jedná o protein akutní fáze, který reaguje na zánět nebo pooperační stav zvýšením své koncentrace v rámci reakce akutní fáze.
Komentář
Příčinu snížení činnosti krvetvorné tkáně nelze často určit. Také vývoj onemocnění lze obtížně odhadnout. Důležité jsou nezralé formy erytrocytů (retikulocyty) a granulocytů (tyčky), které jsou určitým ukazatelem produkce krevních buněk myeloidní řady.
Sérový feritin není plnohodnotou molekulou feritinu, proteinu skladujícího železo intracelulárně. Nitrobuněčný feritin má 24 bílkovinných podjednotek a železo je ukládáno ve středu této makromolekuly. Sérový feritin je určován monoklonální protilátkou, která rozpoznává jednu podjednotku feritinové molekuly. Je to imunoreaktivní feritin v krevní plazmě, který je jen částí feritinové molekuly. Ukázalo se však, že hladina sérového feritinu odráží množství železa v „zásobárnách železa“12 ;např. u menstruujících žen je jeho hladina obecně nižší než u mužů.
121 μg/l sérového feritinu odpovídá asi 8 mg zásobního železa.
Tabulka 1.4 Klasifikace hematologických malignit (zkrácená a zjednodušená verze)
| Myeloidní malignity |
| Myelodysplastický syndrom |
| MDS se specifickou genetickou abnormalitou |
| MDS definovaný morfologicky |
| Akutní myeloidní leukémie (AML) |
| AML se specifickou genetickou abnormalitou |
| AML definovaná stupněm diferenciace |
| Myeloproliferativní onemocnění |
| Chronická myeloidní leukémie (CML) |
| Polycythemia vera |
| Eseciální trombocytémie |
| Primární myelofibróza |
| Mastocytóza |
| Kožní mastocytóza |
| Systémová mastocytóza |
| Lymfoidní malignity |
| Nezralé (prekurzorové) B a T lymfoproliferace |
| B-akutní lymfoblastová leukémie/lymfom (B-ALL, B-LL) |
| T-akutní lymfoblastová leukémie/lymfom (T-ALL, T-LL) |
| Zralé (periferní lymfoproliferace) z B lymfocytů |
| Chronická lymfocytární leukémie (CLL) |
| Difúzní velkobuněčný B-lymfom (DLBCL, z angl. Diffuse large B-cell lymphoma) |
| Folikulární lymfom (FL) |
| Burkittův lymfom (BL) |
| Lymfom z plášťových buněk (MCL, z angl. Mantle cell lymphoma) |
| Hodgkinův lymfom (HL) |
| Malignity z plazmatických buněk a příbuzná onemocnění |
| Monoklonální gamapatie nejistého významu |
| Mnohočetný myelom |
| Lymfoplazmocytární lymfom (LPL) |
| Zralé (periferní) lymfoproliferace z T lymfocytů a NK buněk |
Myeloproliferativní a lymfoproliferativní onemocnění jsou nádorová onemocnění krvetvorné tkáně v její části myeloidní (granulocytopoeza, monocytopoeza, erytropoeza a tvorba trombocytů) nebo lymfoidní (T-, B-buňky). Často se jedná o vznik a expanzi patologického klonu buněk v krvetvorné tkáni. Tento klon pak produkuje většinu nebo část příslušných krevních buněk, které jsou často funkčně méněcenné. Jedná se tedy o patologickou monoklonální hematopoezu.
Obecnou příčinou těchto nemocí jsou genetické změny, zpočátku většinou jedné krvetvorné buňky (somatické mutace). Vznikne „nádorová kmenová buňka“, která je pak zdrojem patologické monoklonální krvetvorby. Následné somatické mutace postihující buňky v tomto buněčném klonu. Krvetvorná tkáň se v organismu nachází v mnoha částech těla a je spojena s krví. Část těchto onemocnění má proto od počátku formu diseminovanou, tj. nemá podobu solidního nádoru.
Patologický klon nejen kvantitativně expanduje oproti klonům normálním, ale často i potlačí produkci krevních buněk z kmenových buněk normálních. Ty však jsou dále přítomny v kostní dřeni. Po terapeutickém potlačení patologického klonu se proto většinou obnoví normální polyklonální krvetvorba – dosáhne se remise, event. úplného uzdravení. Zbytky patologického klonu (hovoří se o „zbytkové nebo reziduální nemoci“) se však po určitém čase mohou opět projevit tvorbou patologických krvetvorných a krevních buněk – dojde k relapsu onemocnění.
!!!začátek petitu!!!
Citlivé metody detekce buněk patřících k patologickému klonu zjišťováním jejich specifického imunofenotypu (pomocí multiparametrové průtokové cytometrie) nebo specifického genotypu (např. pomocí metodi založených na PCR nebo pomocí cíleného sekvenování nové generace) v periferní krvi či v aspirátu kostní dřeně v průběhu léčby či v době remise onemocnění umožňují hodnotit účinnost terapie, prognózu a zejména predikovat relaps onemocnění. V posledních letech se rozvinuly také metody, které jsou schopny detekovat volnou cirkulující nádorovou DNA (ctDNA - z angl. circultating tumor DNA). Předpokládá se, že část nádorových buněk přirozeně podléhá apoptóze a při tomto procesu se z nádorových buněk do cirkulace uvolňuje jejich nádorová DNA. Detekce ctDNA umožňuje sledovat reziduální chorobu také u těch hematologických malignit, u kterých nejsou vyplavovány nádorové buňky do periferní krve či nejsou detekovatelné v kostní dřeni (např. některé typy lymfomů).
!!!konec petitu!!!
Mezi obecné projevy, které se často vyskytují u myeloproliferativních nebo lymfoproliferativních onemocnění, patří únava, opakované a déletrvající subfebrilní a febrilní stavy, častější infekční onemocnění, nadměrné pocení, anémie a s ní související symptomy, trombofilní či naopak krvácivé stavy, ale také hubnutí a kachektizace.
Současná klasifikace hematologických vychází z klinicko-patologických charakteristik jednotlivých onemocnění (Tab. 1.4).
!!!tab. 1.4.!!!
Aplazie (hypoplazie) postihující selektivně tvorbu erytrocytů má formu vrozenou a získanou.
Vrozenou formou je tzv. Diamondův-Blackfanův syndrom.
!!!začátek petitu!!!
Během prvního roku života se vyvine těžká makrocytární anémie s nízkým výskytem retikulocytů a s nedostatkem erytroblastů v kostní dřeni. Onemocnění vykazuje autozomálně dominantní dědičnost, ale značná část případů vzniká sporadicky. U části nemocných se vyskytují ještě jiné vývojové abnormality (např. srdeční vady, rozštěp patra, abnormality palců). Heterozygotní zárodečná mutace v genu RPS19, který kóduje ribozomální protein, je nacházena asi u 25 % případů. U případů s nemutovaným genem RPS19 byly popsány mutace dalších genů, které kódují ribozomální proteiny. U tohoto onemocnění je zvýšené riziko vzniku akutní myeloidní leukémie.
!!!konec petitu!!!
Získaná forma čisté aplazie červené řady (v anglosaské odborné literatuře označovaná zkratkou PRCA – pure red cell aplasia) je charakterizována normochromní normocytární nebo makrocytární anémií a nízkým počtem retikulocytů. V kostní dřeni chybí selektivně erytroidní prekurzory nebo je patrná zástava vývoje na úrovni proerytroblastů. V patogenezi tohoto onemocnění se většinou uplatňuje imunitně zprostředkované potlačení erytropoezy.
!!!začátek petitu!!!
PRCA je většinou idiopatická, v některých případech její výskyt asociuje s některými jinými patologickými stavy: thymom, hematologické malignity, systémové poruchy imunity (systémový lupus erythematodes, revmatoidní artritida), některými virovými onemocněními (HIV, EBV, virové hepatitidy, parvovirus B19 - ), užíváním některých léků.
Čistou aplazii červené řady může také způsobit vytvoření neutralizačních protilátek proti erytropoetinu. Protože je terapeuticky využíván rekombinantní erytropoetin vytvářený na základě lidského erytropoetinového genu, který nevykazuje polymorfismus, jsou tyto případy velmi vzácné.
!!!konec petitu!!!
Fanconiho anémie
Fanconiho anémie (FA) je autozomálně recesivně dědičné onemocnění, jehož podstatou je neschopnost opravit určité typy poškození DNA v důsledku mutací genů, které se podílí na opravě DNA.
!!!začátek petitu!!!
FA má několik fenotypů, z nichž některé se zvýšeně vyskytují u příslušníků určitých etnik. Pro FA je charakteristická přítomnost aplastické anémie s pancytopenií, onemocnění se dále vyznačuje:
Na molekulární úrovni se jedná o velice heterogenní onemocnění a doposud bylo identifikováno 16 genů, jejichž mutace jsou zodpovědné za tento fenotyp.
!!!konec petitu!!!
U jedinců s Fanconiho anémií, kteří se dožijí 40 let věku, se prakticky vždy vyvine aplastická anémie. U části z nich přejde aplastická anémie v myelodysplastický syndrom nebo akutní myeloidní leukémii.
Stavy nedostatku krevních elementů označujeme pojmy anémie (snížené množství hemoglobinu), leukopenie (nedostatek všech „bílých krvinek“), neutropenie (nedostatek neutrofilních granulocytů), lymfopenie (nedostatek lymfocytů) a trombocytopenie (nedostatek trombocytů). Nedostatek všech druhů krevních elementů se označuje jako pancytopenie.
Stavy, kdy je počet krevních elementů zvýšen, se označují jako polycytémie. Zvýšení počtu pouze erytrocytů se označuje také jako polycytémie (též erytrocytóza nebo polyglobulie). Zvýšení počtu různých druhů bílých krvinek se označuje jako leukocytóza. Pojmy jako neutrofilie, eozinofilie a bazofilie jsou používány k označení relativně převažujícího zmnožení neutrofilních, eozinofilních nebo bazofilních granulocytů. Lymfocytóza označuje zmnožení lymfocytů a trombocytémie (nebo trombocytóza) zvýšení počtu trombocytů. V případě zvýšení celkového počtu leukocytů v krvi jsou ve specifických případech používány pojmy leukemoidní reakce a leukémie.
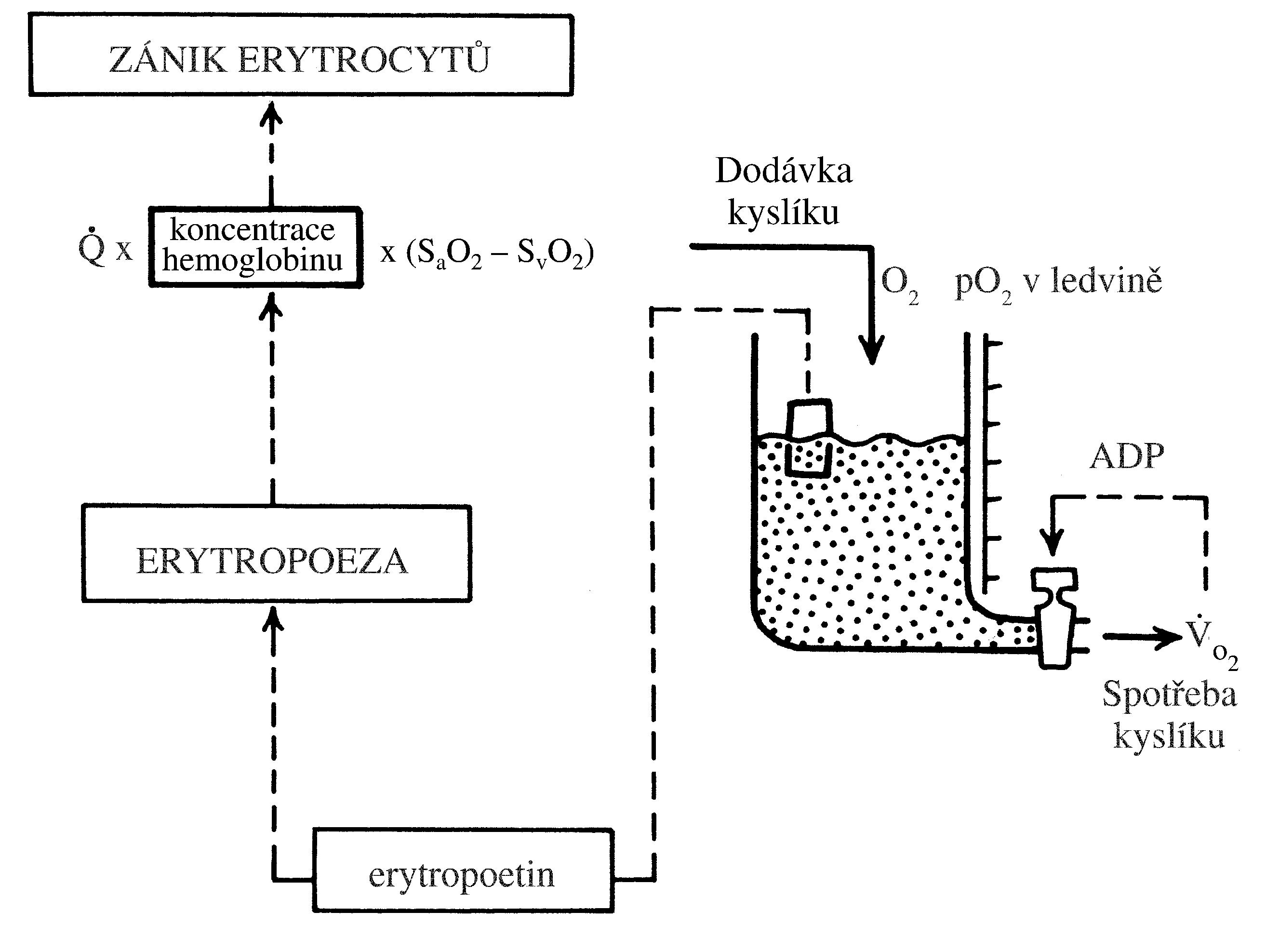
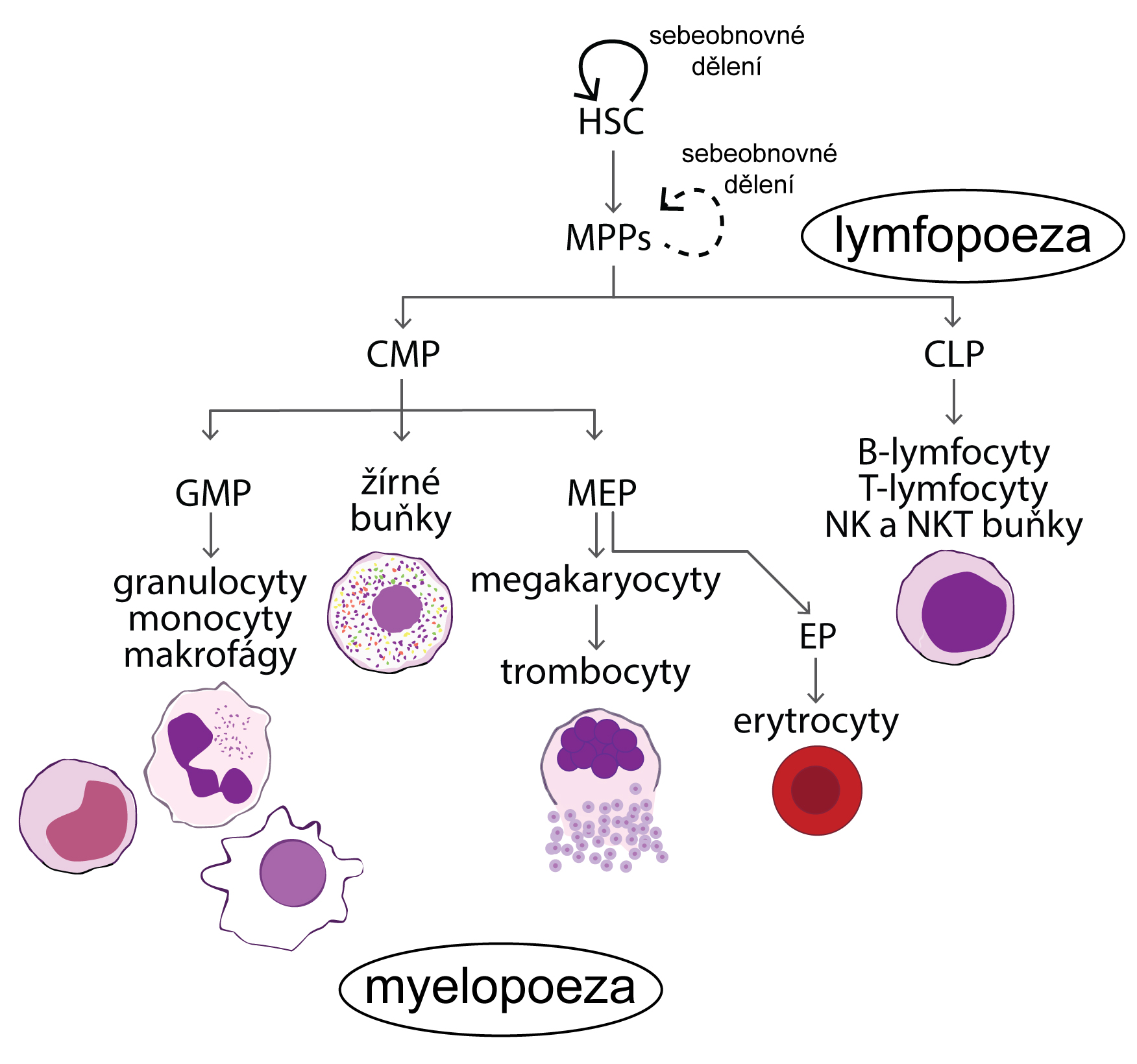
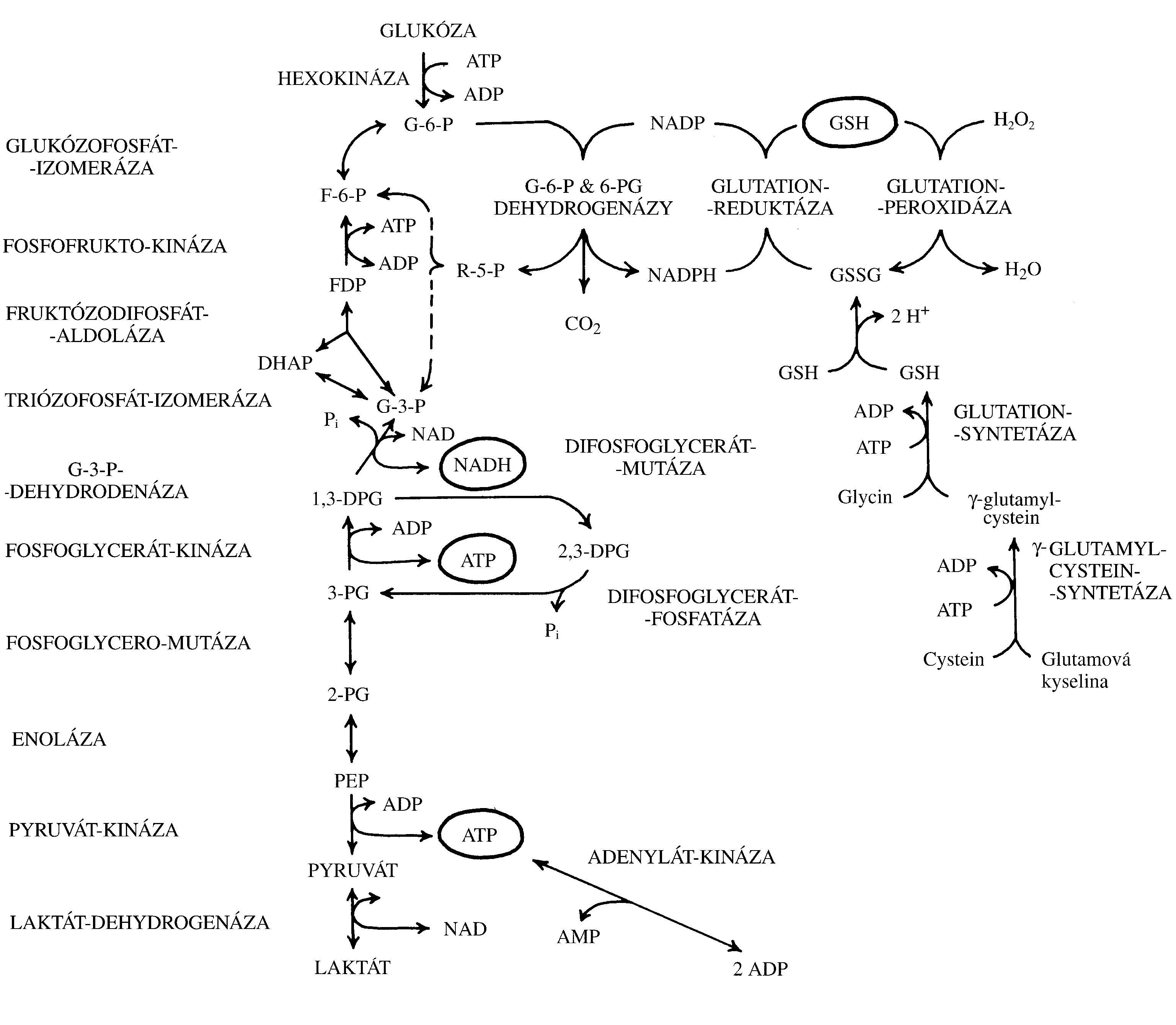
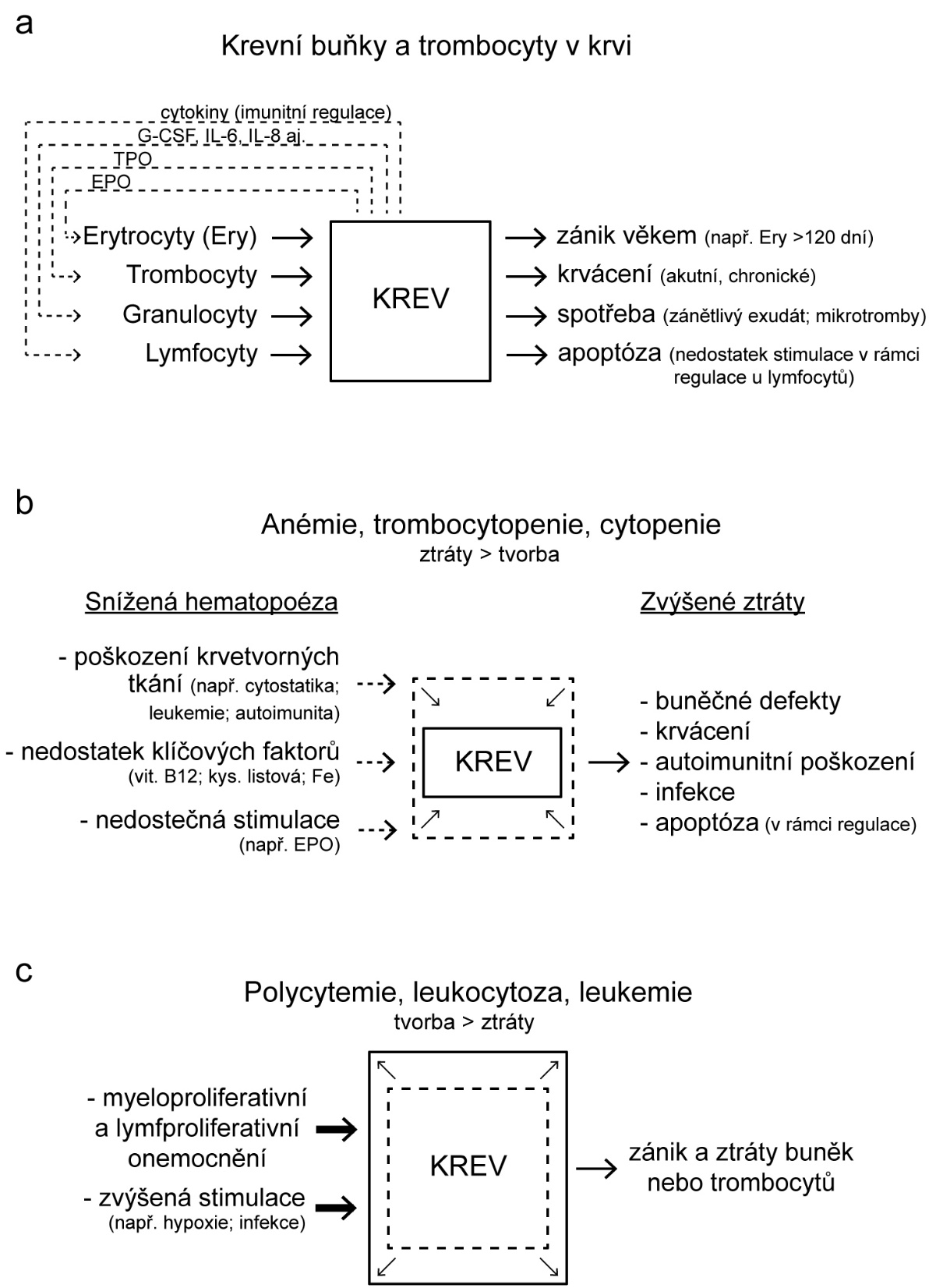
Změny v počtu krevních buněk a krevních elementů (tj. erytrocytů a krevních destiček – dále jen „krevních buněk“) vznikají následkem nerovnováhy mezi zánikem krevních buněk a jejich produkcí (Obr. 1.6). V krvi pak dochází buď k nedostatku, nebo naopak k nadbytku krevních buněk. Nerovnováha mezi zánikem krevních buněk a jejich produkcí může být primárně způsobena buď jejich vystupňovaným zánikem (tj. hemolýzou, spotřebou, krvácením), nebo jejich nedostatečnou/nadměrnou produkcí. V případě trombocytů a granulocytů může být způsobena změna jejich počtu v krvi rovněž změnou jejich sekvestrace ve slezině a v případě granulocytů i jejich uvolněním z marginálního poolu v cirkulaci nebo vyplavením z rezervoáru v kostní dřeni.
Změna počtu krevních elementů může být obecně tím dramatičtější, čím intenzivnější je jejich obrat vzhledem k jejich celkovému počtu v krvi (tj. relativní obrat).
Erytrocyty mají normálně malý obrat, jen 0,8 % za den (žijí 100–120 dní). Často se proto jejich množství v krvi mění pozvolna, třeba po několik týdnů, než změna jejich množství v krvi dosáhne funkčně významného stupně. Pouze v případě velké krevní ztráty nebo intenzivní akutní hemolýzy se může jejich počet prudce snížit.
Granulocyty mají denní obrat velký (až 100 %), jejich počty se proto mohou měnit mnohem dynamičtěji. V případě granulocytů k tomu ještě přispívá skutečnost, že asi polovina granulocytů přítomných v krvi fyziologicky adheruje k endotelové výstelce mikrocirkulace, kde vytváří tzv. marginální pool (hotovost). Granulocyty mohou být z této vazby rychle uvolněny do volného oběhu např. působením adrenalinu. V případě granulocytů existuje ještě jejich další zásoba v kostní dřeni, která převyšuje jejich celkový počet v krvi. Vyplaveny do krve mohou být působením růstového faktoru G-CSF. V tomto případě však zvýšení granulocytů v krvi může být provázeno větším zastoupením jejich forem s nesegmentovaným jádrem, tzv. tyček.
Změny v počtu trombocytů se svojí dynamikou blíží spíše granulocytům než erytrocytům, protože jejich denní obrat je vyšší než 10 % a asi 20–30 % všech trombocytů je uskladněno ve slezině. U splenektomovaných jedinců je proto počet trombocytů v cirkulaci vyšší.
Počet erytrocytů v krvi je asi 1000krát vyšší než počet granulocytů (ty představují asi 70 % ze všech leukocytů) a asi 20krát vyšší než počet trombocytů. Krvácení spojené se ztrátou krve proto významně sníží především počet erytrocytů (také koncentraci hemoglobinu v krvi a hodnotu hematokritu). Počty granulocytů a trombocytů se také rychleji doplní z důvodu jejich relativně vysoké denní produkce. Proto je akutní, jakož i chronické krvácení spojeno se vznikem anémie a ne leukopenie a trombocytopenie.
!!!obr. 1.6.!!!
11Anémie, snížený počet erytrocytů v krvi a snížený hematokrit, zvýrazňují procentuální podíl retikulocytů mezi erytrocyty, a nadhodnocují proto odhad produkce erytrocytů dělaný na základě stanovení procentního podílu retikulocytů. Proto je potřebná tato korekce.
Tabulka 1.9 Morfologická klasifikace anémií
Normocytární anémie (průměrný objem erytrocytů, MCV, je 80–95 fl)
Makrocytární anémie (průměrný objem erytrocytů, MCV, je větší než 95 fl)
Mikrocytární anémie (průměrný objem erytrocytů, MCV, je menší než 80 fl)
Normochromní anémie (průměrná koncentrace hemoglobinu v erytrocytech, MCHC, je 300–350 g/l erytromasy)
Hypochromní anémie (průměrná koncentrace hemoglobinu v erytrocytech, MCHC, je méně než 300 g/l erytromasy)
14Retikulocyty dozrávají v krvi v erytrocyty během jednoho až dvou dnů. Proto je normální zastoupení retikulocytů v krvi asi 1–1,5 %. Při zvýšené stimulaci krvetvorné tkáně erytropoetinem jsou do cirkulace vyplavovány méně zralé retikulocyty, které zůstávají ve stadiu retikulocytu déle než dva dny. Výskyt retikulocytů v krvi pak nadhodnocuje odhad intenzity jejich produkce (erytropoezy). Proto je výskyt retikulocytů v krvi jen orientačním ukazatelem erytropoetické aktivity krvetvorné tkáně.
Idiopatické a sekundární aplastické anémie
Aplastická anémie (AA) je získané onemocnění, které je charakterizováno výrazným snížením buněčnosti kostní dřeně a neschopností produkovat zralé krevní elementy v potřebnéém množství, což působí anémii, trombocytopenii, neutropenií či pancytopenii. Při aplastické anémii je v kostní dřeni nedostatek erytroblastů, megakaryocytů a prekurzorů granulocytů. Kostní dřeň je hypocelulární a chybí zde nebo jsou silně redukovány buňky CD34+, které obsahují buňky kmenové a progenitory.
AA lze dělit na:
Výzanmným patogenetickým mechanismem je porucha hematopoetických kmenových buněk, která redukuje její schopnost proliferace, sebeobnovy a diferenciace.
!!!začátek petitu!!!
Mutace v genech TERC a TERT, které kódují komponenty telomerázového komplexu jsou popisovány asi u 5-10 % pacientů s primární AA (telomerázy zabraňují zkracování chromozomálních konců, tzv. telomér, ke kterému přirozeně dochází při buněčném dělení). Tyto mutace způsobují nízkou aktivitu telomerázového komplexu a předčasný zánik hematopoetických kmenových buněk.
!!!konec petitu!!!
Někdy dojde v hematopoetických buňkých k expresi neoantigenů, které vyvolají imunitní odpověď a poškození hematopoezy cytotoxickými T lymfocyty. Část pacientů s AA odpovídá na imunosupresivní léčbu anti-thymocytárním globulinem. Další léčebnou možností je alogenní transplantace krvetvorných buněk. Nutná je také intenzivní podpůrná terapie spočívající v opakovaných transfúzích erytrocytů, trombocytů či anti-infekční profylaxi.
Sekundární AA zpsobené cytotoxickým působením cytostatik nebo ionizujícím zářením jsou přechodné a upraví se spontánně po ukončení působení patogenní noxy.
Klinická manifestace je odvozena od anémie (bledost, únava, zhoršení mentálních funkcí, tachykardie, hypercirkulační stav, systolický šelest) a trombocytopenie (krvácivé projevy – petechie, hematomy, krvácení z dásní, krvácení do trávicího ústrojí, menoragie a metroragie). Dále je zvýšené riziko výskytu bakteriálních a mykotických infekcí při neutropenii. V periferní krvi je také velmi málo retikulocytů.
!!!začátek petitu!!!
AA dělíme dle nálezu v kostní dřeni (% buněčnosti) a absolutních počtů krevních elementů v periferní krvi na těžkou a středně těžkou AA. U těžké AA je buněčnost kostní dřeně snížena pod 30% a počty krevních elementů alespoň ve 2 ze 3 řad jsou významně sníženy (počet neutrofilních granulocytů v krvi snížen pod 5 x 108/l (500/μl), je přítomna závislost na transfúzích s počtem trombocytů pod 2 x 1010/l (20 000/μl) a retikulocytů pod 6 x 1010/l (60 000/μl)11.
!!!konec petitu!!!
Fanconiho anémie (FA) je autozomálně recesivně dědičné onemocnění, jehož podstatou je neschopnost opravit určité typy poškození DNA v důsledku mutací genů, které se podílí na opravě DNA.
!!!začátek petitu!!!
FA má několik fenotypů, z nichž některé se zvýšeně vyskytují u příslušníků určitých etnik. Pro FA je charakteristická přítomnost aplastické anémie s pancytopenií, onemocnění se dále vyznačuje:
Na molekulární úrovni se jedná o velice heterogenní onemocnění a doposud bylo identifikováno 16 genů, jejichž mutace jsou zodpovědné za tento fenotyp.
!!!konec petitu!!!
U jedinců s Fanconiho anémií, kteří se dožijí 40 let věku, se prakticky vždy vyvine aplastická anémie. U části z nich přejde aplastická anémie v myelodysplastický syndrom nebo akutní myeloidní leukémii.
Obr.1.12 Účast hemoglobinu v transportu kyslíku krví. Koncentrace hemoglobinu závisí na intenzitě zániku erytrocytů a na jejich doplňování erytropoezou. Erytropoeza je řízena erytropoetinem, který je produkován v závislosti na tlaku kyslíku ve specifických buňkách ledviny
2Hodnota viskozity krve η je obsažena v čitateli matematického výrazu, který charakterizuje odpor cévního řečiště proudění krve: R = 8ηd/πr4.
Tabulka 1.10 Patofyziologická klasifikace anémií
Anémie ze zvýšených ztrát erytrocytů
Anémie ze snížené produkce erytrocytů
Na obrázku 1.12 je znázorněno začlenění koncentrace hemoglobinu do transportu kyslíku a regulace koncentrace hemoglobinu v krvi mechanismem, který ovlivňuje množství vytvářených erytrocytů krvetvornou tkání v závislosti na její stimulaci erytropoetinem. Na obrázku 1.13 jsou ukázány hladiny erytropoetinu ve vztahu ke koncentraci hemoglobinu v krvi. Erytrocyty přirozeně zanikají asi po 120 dnech (během této doby asi dvěstětisíckrát přenesou kyslík z plic do tkání). Počet erytrocytů a koncentrace hemoglobinu v krvi jsou výsledkem rovnováhy mezi zánikem a tvorbou erytrocytů. Denně zaniká asi 0,8 % erytrocytů ze všech erytrocytů přítomných v krvi, tj. asi 2 x 1011 erytrocytů (přibližně 20 ml erytromasy). Stejné množství musí být dodáno krvetvornou tkání do krve ve formě retikulocytů14. Anémie vznikne následkem nerovnováhy mezi zánikem (ztrátou) erytrocytů a jejich produkcí, nerovnováhy posunuté směrem k vyššímu zániku. Z toho vyplývá, že existují dva patogenetické mechanismy vzniku anémie:
- nedostatečná tvorba erytrocytů
- zvýšení ztrát erytrocytů (hemolýzou nebo krvácením).
!!!obr. 12!!!
!!!obr. 13!!!
Anémii může způsobovat buď jen jeden z těchto dvou mechanismů, nebo se mohou na vzniku anémie účastnit oba (rychlost rozvoje anémie a její stupeň se tím zvýší). Na druhé straně může být zvýšený zánik erytrocytů částečně nebo i zcela kompenzován zvýšenou produkcí erytrocytů. Je-li kompenzace účinná, označuje se takov stav jako kompenzovaný hemolytický syndrom.
Poslední obecná úvaha upozorňuje na skutečnost, že každá změna v intenzitě zániku erytrocytů nebo v jejich produkci spěje samovolně k ustálení nového rovnovážného stavu z hlediska celkového množství cirkulujících erytrocytů.
!!!začátek petitu!!!
Ozřejměme si to na příkladu teoretického poklesu produkce erytrocytů na jednu polovinu, tj. na 1 x 1011/den. Množství zanikajících erytrocytů (2 x 1011/den) bude zpočátku převažovat nad jejich produkcí. Jak se však bude v cirkulaci snižovat celkový počet erytrocytů, bude se snižovat i absolutní hodnota zanikajících erytrocytů, která bude stále 0,8 % z aktuálního množství. Po snížení celkového počtu erytrocytů na polovinu se ustaví nová rovnováha mezi zánikem a produkcí (1 x 1011/24 h zaniká a vzniká) a anémie se již dále nebude prohlubovat.
!!!konec petitu!!!
Tradiční morfologická klasifikace anémií vychází z normálního nebo změněného fenotypu erytrocytů v krvi. Patofyziologie rozděluje anémie na anémie vznikající ze zvýšených ztrát erytrocytů a na anémie vznikající z nedostatečné produkce erytrocytů.
Morfologická klasifikace anémií vychází z krevního obrazu, stanovení počtu erytrocytů, jejich velikosti, jejich hemoglobinizace a případně jejich patologických tvarů. Tyto ukazatele slouží k počáteční orientaci při zjišťování příčiny vzniku anémie.
Jako normocytární, normochromní anémie se označují ty stavy, kdy je snížená hladina hemoglobinu v krvi, je snížený počet erytrocytů v krvi, ale tyto se svou velikostí (MCV – mean cell volume), obsahem hemoglobinu (MCH – mean cell hemoglobin) a průměrnou koncentrací hemoglobinu (MCHC – mean cell hemoglobin concentration) neliší od erytrocytů zdravých jedinců (MCV = 80–95 fl).
Jako makrocytární anémie se označují ty anémie, při kterých je průměrný objem erytrocytů (MCV) větší než 95 fl.
Jako mikrocytární anémie se označují ty anémie, při kterých je průměrný objem erytrocytů (MCV) pod 80 fl. Mohou být normochromní, tj obsah hemoglobinu je v nich snížen (pod 26 pg) jen z důvodu jejich menšího objemu, nebo hypochromní, kdy je jejich obsah hemoglobinu snížen nejen z důvodu mikrocytózy, ale i jejich nedostatečné hemoglobinizace (i průměrná koncentrace hemoglobinu, MCHC, je v nich pak snížena pod 0,32).
Distribuční šíře velikosti erytrocytů (RDW – red-cell distribution width) je ukazatelem možné anizocytózy. Tabulka 1.9 ukazuje, jak je morfologická klasifikace používána jako pomocné diagnostické kritérium.
!!!tab. 1.9.!!!
Patofyziologická klasifikace anémií rozlišuje, zda je příčinou anémie nedostatečná produkce erytrocytů nebo jejich zvýšené ztráty (Tabulka 1.10).
!!tab. 1.10.!!!
Symptomy provázející onemocnění krve a krvetvorného systému mohou být natolik nespecifické a nevýrazné, že onemocnění může být zjištěno až na základě rutinního laboratorního vyšetření krevního obrazu, prováděného z jiných důvodů. Symptomy upozorňující na poruchy funkce krve a krvetvorby mohou být krvácivé projevy (méně často tromboembolie), časté infekce, zvýšené teploty, hubnutí, pocení, únava, bledost kůže a sliznic, námahová dušnost.
Myeloftiza označuje náhradu kostní dřeně vazivovou tkání v reakci např. na invazi nádorových buněk do kostní dřeně (např. u metastatického karcinomu prsu, plic nebo prostaty). Přítomnost nádorových buněk v kostní dřeni vede ke stimulaci fibroblastů, depozici kolagenu s výslednou fibrotizací kostní dřeně. Normální krvetvorba je ultačena. V periferní krvi se vyskutuje anémie, často doprovázená trombocytopenií nebo leukopenií. Komplexní změny mikroprostředí kostní dřeně vedou k vyplavování nezralých krevních elementů do periferní krve. Na rozdíl od idiopatické myelofibrózy jde při myeloftize o sekundární proces, reakci na primární onemocnění.
13To způsobuje náchylnost ke vzniku různých malignit a je základem depoxybutanového testu, který je pro Fanconiho anémii vysoce citlivý a specifický (v obou případech z 99 %).
Proudění krve v cévním řečišti je hydrodynamický děj, který je ovlivňován mnoha faktory. Je určován nejen přítomností buněk, ale i různým charakterem cévního řečiště v jeho jednotlivých částech, jeho větvením, vnitřním průměrem, poddajností, patologickými změnami, jako jsou zúžení, uzávěr, změna endotelií, přítomnost fibrinových vláken, snížení nebo zvýšení poddajnosti cévní stěny aj. Vlastnosti krve jako tekutiny, která neustále proudí cévním systémem, jsou dány jednak vlastnostmi plazmy a jednak množstvím a vlastnostmi krevních buněk, jejich plasticitou – deformabilitou.
Častou příčinou změn reologických vlastností krve jsou změny hematokritu při patologických stavech označovaných jako polycytémie a anémie. Polycytémie je jednou z příčin hyperviskózního syndromu, při kterém se zvýšení viskozity krve v cirkulaci projevuje zpomalením proudění krve. Při stejném tlakovém gradientu se zvýšení viskozity krve projeví stejně jako zvýšení odporu cévního řečiště proudění krve2. Zvýšení viskozity krve proto predisponuje ke zvýšení krevního tlaku, které je potřebné pro udržení dostatečné perfúze tkání. Zpomalení proudění krve dále zvyšuje koagulabilitu krve, protože lokálně aktivované koagulační faktory jsou pomaleji odplavovány z místa svého vzniku. Hyperviskózní syndrom tím může vedle chronicky zvýšených nároků na srdeční práci představovat i akutní ohrožení života3. Hyperviskózní syndrom může být také způsoben velkým zvýšením počtu leukocytů v krvi při leukémii nebo zvýšením koncentrace plazmatických bílkovin.
Snížení viskozity krve při anémii ovlivňuje cirkulaci krve méně než polycytémieSnížení viskozity krve se projeví jako „snížení periferního odporu cévního řečiště“. Tendence k poklesu krevního tlaku způsobuje hyperkinetickou cirkulaci, zvýšení srdečního výdeje. To se projeví zvláště v období zvýšené fyzické aktivity, protože v důsledku vazodilatace v pracujících svalech dojde k dalšímu poklesu periferního odporu cirkulace.
Změněné vlastnosti erytrocytů nebo aktivace granulocytů, monocytů a endotelu zánětovými cytokiny mohou také významně změnit schopnost krve proudit cévami o malém vnitřním průměru. Při onemocnění srpkovitou anémií nebo při infekci erytrocytů parazity Plasmodium malariae, lokální ischémie v různých tkáních může být dominantní patofyziologickou poruchou těchto onemocnění projevujíví se intenzivní bolestí a závažným poškození orgánů.
Obr.1.13 Vztah mezi koncentrací hemoglobinu v krvi a hladinou erytropoetinu v plazmě. K – kontrolní zdravé osoby; 1 – nemocní s anémií, která není způsobena nedostatečnou tvorbou erytropoetinu; 2 – nemocní s anémií, která je způsobena nedostatečnou tvorbou erytropoetinu; 3 – nemocní s pravou polycytémií; 4 – nemocní se „sekundární“ polycytémií způsobenou celkovou hypoxií organismu
3Injekcemi erytropoetinu lze zvýšit hematokrit, a tím vazebnou kapacitu krve pro kyslík. Při sportovní zátěži spojené s dehydratací organismu se může hematokrit, a tedy i viskozita krve dále zvýšit. Komplikacemi hyperviskózního syndromu (srdeční selhání nebo tromboembolie) jsou vysvětlována některá náhlá úmrtí sportovců, u kterých se předpokládalo použití erytropoetinu pro zvýšení aerobní kapacity organismu.
Krev je součástí všech tkání, orgánů a orgánových systémů. Část její patofyziologie je proto probírána u některých obecných patofyziologických stavů (např. v rámci hypoxie, poruch acidobazické rovnováhy). Svým objemem a reologickými vlastnostmi může krev významně ovlivňovat činnost cirkulačního aparátu. Celkový objem krve, ve vztahu k vnitřnímu objemu cirkulačního aparátu, je charakterizován jako normovolémie, hypovolémie nebo hypervolémie. Většina proteinů přítomných v krevní plazmě je syntetizována a do plazmy secernována játry. Patofyziologie krve by měla zahrnout všechny tyto souvislosti. Klinický obor hematologie se převážně zabývá nemocemi buněčné části krve a poruchami funkce krvetvorných tkání, ale také v plazmě přítomnými prokoagulačními a antikoagulačními faktory.
Tyto hematologické syndromy jsou určitým protipólem myeloproliferativních a lymfoproliferativních syndromů v tom smyslu, že krvetvorná tkáň obsahuje málo prekurzorů krevních buněk, je hypoplastická. Většinou se jedná o následky primární poruchy nebo poškození krvetvorných kmenových a progenitorových buněk, mnohem vzácněji o následky poškození stromatu krvetvorné tkáně. Hyperproliferativní syndromy mohou někdy přejít v aplastický syndrom, a naopak některé aplastické syndromy mohou přejít v některou formu akutní myeloidní leukémie. Ukazuje to na biologickou poruchu krvetvorby na úrovni jejich kmenových buněk.
Obrázek 1.14: Porucha metabolismu železa u anémie chronických chorob (legenda v textu).
Erytropoetin je esenciálním faktorem pro erytropoezu. V krvi je malý počet retikulocytů. Erytropoetin je produkován peritubulárními buňkami kortexu ledvin. Nedostatek erytropoetinu vzniká především u oboustranně nefrektomovaných jedinců nebo u nemocných se závažným chronickým poškozením obou ledvin (viz Obr. 1.10). U oboustranně nefrektomovaných a dialyzovaných pacientů se anémie většinou ustálí na hodnotách hematokritu mezi 25 a 15 %. Anémie je normocytová a normochromní. Určitá produkce erytrocytů a erytropoetinu je zachována a přičítá se produkci erytropoetinu jaterní tkání.
Anémie chronických chorobAnémie provází některá chronická zánětlivá onemocnění, např. autoimunitní onemocnění (revmatoidní artritida), chronické infekce nebo onemocnění nádorová. Na jejím vzniku se podílí několik patogenetických mechanismů. Důležitou roli hraje zvýšení hladiny prozánětových cytokinů TNFα, IL-1, IL-6 nebo IFN-γ. Ty ovlivňují řadu dějů.
Porucha metabolismu železa je klíčová v patogenezi anémie chronických chorob.
!!!začátek petitu!!!
Prozánětlivé cytokiny, zejména IL-6 způsobí zvýšení hladiny peptidu hepcidinu. Hepcidin je klíčový regulátor metabolismu železa (Obr. 1.14) a tvoří se převážně v játrech. Hepcidin se váže na feroportin (transmembránový protein potřebný pro export železa z makrofágů nebo enterocytů), působí jeho degradaci, a to vede ke snížení absorbce železa enterocyty tenkého střeva a fixaci zásobního železa v makrofázích. Výsledkem je snížení hladiny plazmatického železa a jeho nedostatek pro erytropoezu
!!!konec petitu!!!
!!!obr. 1.14!!!
Působením prozánětlivých cytokinů dále dochází ke snížení proliferace erytroidních prekurzorů v kostní dřeni, k nižší tvorbě erytropoetinu, než by odpovídalo stupni tkáňové hypoxie, a k mírně zkrácenému přežívání erytrocytů.
Anémie doprovázející chronická onemocnění bývá mírného až středního stupně, hemoglobin bývá většinou v rozmezí 70–110g/l. Anémie bývá normochromní, normocytární, při delším trvání onemocnění může být těžšího stupně s hypochromií a mikrocytózou. Laboratorně nacházíme normální nebo snížené hladiny sérového železa, normální nebo snížené množství transferinu, normální nebo sníženou celkovou vazebnou kapacitu pro železo a normální nebo sníženou saturaci transferinu. Hladina sérového ferritinu (odrážející množství zásobního železa) je však normální nebo zvýšená. Léčba anémie doprovázející chronická onemocnění by měla směřovat primárně k léčbě základního onemocnění.
Obrázek 1.4 Kolonie buněk (klony) z jedné progenitorové buňky. BFU-E časný erytroidní progenitor, CFU-E – pozdní erytroidní progenitor blízký proerytroblastu; CFU-G a CFU-M – progenitory pro granulocyty a monocyty/makrofágy
Hematopoetické (krvetvorné) kmenové buňky představují vrchol hierarchického uspořádání buněk krvetvorby (obr. 1.1). Za hematopoetickou (krvetvornou) kmenovou buňku je považována buňka, která je schopna obnovit veškerou krvetvorbu po jejím úplném zničení a tuto krvetvorbu dlouhodobě udržovat. Základními charakteristikami HSC je tedy 1. multipotence, tj. schopnost diferencovat do několika typů krevních buněk a 2. schopnost sebeobnovy, tj. schopnost udržovat a obnovit svoji vlastní populaci.
Kmenové buňky jsou málo početné a představují asi 1 na 105 buněk kostní dřeně. Za normálních okolností se zřídka dělí. Dělit se mohou dvěma způsoby. Tzv. symetrické dělění dá vznik dvěma stejným dceřiným buňkám, dvěma buňkám kmenovým nebo dvěma buňkám progenitorovým. Asymetrické dělení dá vznik jedné buňce kmenové a jedné buňce, která se diferencuje na buňku progenitorovou. Asymetrické dělení představuje způsob, jak zachovat populaci buněk kmenových a zároveň umožnit produkci zralých krevníh elementů. Kmené buňky se mohou uvolnit do krve, odkud je možné je získat za účelem jejich transplantace.
Buňky progenitorové jsou hierarchicky uspořádány. Vznikají diferenciací z HSC a postupně ztrácejí schopnost sebeobnovy a nabývají specifického vývojového potenciálu. Mohou dát vznik omezenému počtu buněk (Obr. 1.4). Jen některé lze transplantovat, ale udržují potom tvorbu příslušných buněk nanejvýše několik týdnů. Různě itenzivně proliferují a jejich dělení převážně zajišťuje obněnu krátkodobě žijících krevních buněk. Stejně jako buňky kmenové nelze ani buňky progenitorové rozeznat mikroskopem. Jsou podobné lymfocytům.
!!!obr. 1.4.!!!
Prekurzory krevních buněk jsou vývojová stadia krevních buněk přítomných v krvetvorné tkáni, které lze rozpoznat a klasifikovat podle jejich specifických morfologických, histochemických a antigenních znaků (Obr. 1.5).
!!!obr. 1.5.!!!
Protože jedna kmenová buňka se může stát zdrojem všech buněčných typů krevních buněk, produkovaných v potřebném množství a vzájemných poměrech, můžeme se setkat se stavy tzv. monoklonální krvetvorby. Většinou se však v těchto případech jedná o patologickou krvetvorbu (viz dále). Normální krvetvorba je polyklonální, protože v určitém stadiu časné embryogeneze vznikne paralelně několik kmenových buněk, které jsou zdrojem krevních buněk po celý život. Každá z těchto kmenových buněk je zdrojem specifického klonu krevních buněk (u žen se mohou buňky příslušné k jednotlivým klonům lišit např. inaktivací otcovského nebo mateřského chromozomu X).
Krvetvorná tkáň nemá zdánlivě pevnou strukturu. Stroma krvetvorné tkáně je tvořeno řadou buněčných typů, proteiny extracelulární matrix, růstovými a regulačními faktory. Významnou roli má i specifická mikrocirkulace krvetvorné tkáně se sinusoidy zvýšeně prostupnými pro krevní buňky. Krvetvorné kmenové buňky jsou v kostní dřeni ve specifických místech, označovaných jako „niche“, v blízkosti endosteálního povrchu a také v blízkosti endotelu sinusoid. Jsou v blízkosti osteoblastů, endotelu, pericytů, fibrocytů, makrofágů, megaklaryocytů, mezenchymových kmenových buněk, adipocytů, které jsou zdrojem cytokinů a růstových faktorů, které řídí jejich činnost (Obr. 1.3).
Z lékařského hlediska je významnou vlastností hematopoetického stromatu jeho relativní odolnost vůči některým noxám, vůči kterým jsou naopak krvetvorné kmenové buňky značně citlivé. Jedná se o ionizující záření a protinádorové léky ze skupiny cytostatik. Při léčebném použití těchto léků je stroma krvetvorné tkáně podstatně méně poškozeno než vlastní krvetvorba. To je důležitý předpoklad pro regeneraci krvetvorby z přeživších nebo transplantovaných kmenových, která může být rychlá a intenzivní. Mezi buňkami stromatu kostní dřeně jsou i mezenchymové kmenové buňky. Ty se mohou diferencovat ve fibrocyty a adipocyty, chondrocyty a osteoblasty i jiné druhy buněk.
!!!obr. 1.3.!!!
Obrázek 1.3: Schéma niche. Kmenové buňky (HSC) se nacházejí v blízkosti kapilár (sinusoid) a v blízkosti endosteálního povrchu kosti. Jsou v kontaktu s řadou buněk a cytokinů. CXCR4/CXCL12 a c-kit/SCF jsou páry receptorů a jejich ligandů (cytokinů). LepR+ buňky produkují stem cell faktor (SCF) a cytokin CXCL12.
Do této kategorie patří již probrané anémie aplastické, které souvisí s poškozením kmenových buněk. Dále sem patří anémie způsobená nízkou produkcí erytropoetinu a anémie vznikající v důsledku nedostatku železa, kyseliny listové a vitaminu B12, anémie sideroblastové a anémie dyserytropoetické.
Tab. 1.11 Laboratorní ukazatele sideropenie.
| Sérový feritin(ng/ml) | Saturace transferinu(%) | Volný protoporfyrin v erytrocytech(μg/100 ml erytrocytů) | Koncentrace hemoglobinu v krvi(g/l) | |
| Normální stav | > 60 | 35 | 30 | > 120 |
| Prelatentní sideropenie | < 12 | 35 | 30 | > 120 |
| Latentní sideropenie | < 12 | < 15 | > 100 | > 120 |
| Manifestní sideropenie | < 12 | < 15 | > 100 | < 120anémie |
Obrázek 1.5 Změna fenotypu krvetvorné buňky, která obsahuje produkt fúzního genu abl-bcr
Při normální erytropoeze, kdy se za 24 hodin vytvoří asi 2 x 1011 erytrocytů (toto množství je obsaženo v asi 40 ml krve), je pro syntézu hemoglobinu zapotřebí asi 20–25 mg železa. Nedostatek železa zpomalí vyzrávání erytroblastů, jehož součástí je jejich hemoglobinizace. Erytroblasty déle setrvávají v kostní dřeni. Přestože je denní produkce erytrocytů snížená a v krvi je málo retikulocytů, v kostní dřeni je normální nebo dokonce zvýšené množství pomalu zrajících erytroblastů. Protože zrání erytroblastů do stadia ortochromního erytoblastu (kdy dochází k enukleaci jádra a jeho změně v retikulocyt) je prodloužené, mohou erytroblasty projít větším počtem mitotických dělení. To vede ke zmenšení jejich objemu a jejich průměru (mikrocyty). Při nedostatku železa značného stupně nejsou tyto mikrocyty dostatečně hemoglobinizovány, jsou hypochromní. Plně rozvinutá anémie z nedostatku železa je proto mikrocytární a hypochromní.
!!!začátek petitu!!!
Nedostatek železa se většinou rozvíjí postupně. Mezi laboratorní nálezy, které předcházejí vzniku zjevné mikrocytárníé hypochromní anémie, patří snížení hladiny sérového feritinu (vyčerpání zásob železa), snížení saturace transferinu železem (normální saturace je 30–50 %), zvýšení koncentrace transferinu v plazmě a tomu odpovídající zvýšení celkové vazebné kapacity plazmy pro železo. Celkové množství železa obsaženého v jednotkovém objemu krve se sníží. V kostní dřeni nejsou přítomny „buňky barvitelné na železo“ (siderocyty). Velikost erytrocytů se začne více lišit a zvětší se hodnota udávající distribuční šíři velikosti erytrocytů (RDW). Tomu v krevním nátěru odpovídá anizocytóza16, při těžkém nedostatku železa i poikilocytóza17. V erytrocytech je zvýšené množství volného protoporfyrinu (normálně asi 0,5 μmol/l a při nedostatku železa nad 1,8 μmol/l). Je v nich také zvýšena koncentrace Zn-protoporfyrinu, protože ferochelatáza, poslední enzym hemové syntézy, zvýšeně inkorporuje zinek do protoporfyrinu místo nedostatkového železa. Laboratorní vyšetření je pro diagnózu anémie z nedostatku železa klíčové.
!!!konec petitu!!!
Pomocí laboratorního vyšetření lze rozlišit tři stupně nedostatku železa (Tab. 1.11):
* nedostatečné zásoby železa (prelatentní sideropenie),
* erytropoeza uskutečňovaná při nedostatku železa (latentní sideropenie),
* anémie z nedostatku železa (manifestní sideropenie).
!!!tab. 1.11.!!!
Železo je vedle syntézy hemoglobinu (a jiných hemoproteinů) významné pro buněčné dělení, protože je obsaženo v reduktáze ribonukleotidů, enzymu nezbytném pro tvorbu deoxynukleotidů potřebných pro syntézu DNA. Nedostatek železa se proto projevuje také změnami na sliznicích a kožních adnexech. Ty jsou zdrojem poměrně nespecifických příznaků, jako jsou ragády ústních koutků, obtíže při polykání a změny nehtů, jejich změknutí a kroucení.
Obr. 1.15 Úloha kyseliny listové (folátů) a vitaminu B12 v syntéze DNA. THF – tetrahydrofolát, dUMP – deoxyuridin-monofosfát, dTMP – deoxytymidin-monofosfát, dTTP – deoxytymidin-trifosfát
Zásoby kyseliny listové v těle se odhadují na 5–20 mg, při její minimální potřebě asi 0,05 mg/den. Její nedostatek se proto může projevit během týdnů až měsíců po vzniklé negativní bilanci mezi mezi příjmem a spotřebou kyseliny listové. Hlavním zdrojem kyseliny listové je zelenina. Některé formy kyseliny listové jsou citlivé k vyšší teplotě při eventuální úpravě zeleniny. Nedostatek z nutričních důvodů je popisován především u alkoholiků, drogově závislých jedinců a starých lidí. Porucha vstřebávání v proximálním jejunu provází tropickou sprue, ale může provázet i glutenovou enteropatii (netropickou sprue).
Kyselina listová se v organismu nadměrně spotřebovává tehdy, dochází-li ve tkáních k intenzivnímu buněčnému dělení. Příklady zvýšené spotřeby kyseliny listové proto zahrnují: období rychlého růstu v dětství a v pubertě, vývoj plodu během těhotenství, nádorové onemocnění, intenzivní hemolytickou anémii s kompenzací a některá kožní onemocnění s intenzivní tvorbou epidermis. Ke zvýšeným ztrátám kyseliny listové může docházet u chronicky hemodialyzovaných pacientů. Poměrně častou příčinou projevů nedostatku kyseliny listové je léčba protinádorovým a imunosupresivním lékem – metotrexátem. Je klinickou zkušeností, že se za těchto stavů mohou objevit známky megaloblastické, makrocytární anémie.
Metotrexát je silným inhibitorem enzymu dihydrofolátreduktázy. Tento enzym umožňuje reutilizaci tetrahydrolistové kyseliny použité při syntéze deoxytymidinmonofosfátu. Existují i další protinádorové léky, jejichž hlavní mechanismus účinku spočívá v inhibici tohoto enzymu.
16Erytrocyty obvyklého tvaru, ale nestejně velké.
17Erytrocyty nestejně velké a tvarově změněné.
Anémie je definována jako snížení koncentrace hemoglobinu v krvi, většinou i počtu erytrocytů v krvi a hodnoty hematokritu. Za kritickou hodnotu je považována koncentrace hemoglobinu, a to pro její úlohu v transportu kyslíku. Anémie je symptom, který se vyskytuje u některých patologických stavů. Jako diluční nebo relativní anémie se označuje stav, kdy anémie je způsobena zvětšeným objemem plazmy. Anémie je symptom vyskytující se u řady nemocí.
!!!začátek petitu!!!
Při mírných anémiích není vždy snadné rozhodnout o existenci anémie jako o patologickém stavu. Koncentrace hemoglobinu v krvi se mezi zdravými jedinci může lišit o 10 i 20 %, je individuálně rozdílná.
!!!konec petitu!!!
Při anémii je normální parciální tlak kyslíku v arteriální krvi, hemoglobin je v arteriální krvi plně saturován kyslíkem, ale protože je ho málo, snižuje se obsah kyslíku v arteriální krvi. Následkem toho v krvi klesá více parciální tlak kyslíku při jejím průchodu tkáňovými kapilárami (obr. 1.11). Ve venózní krvi je proto nejen snížen obsah kyslíku, ale i jeho parciální tlak.
!!!obr. 1.11.!!!
Soubor symptomů, které často provázejí snížení koncentrace hemoglobinu v krvi, lze označit za anemický syndrom. Patří sem bledost sliznic a kůže, únava, pokles tělesné výkonnosti (zvláště vykonávané maximální intenzitou), zadýchávání při námaze, tachykardie (zvláště při námaze). Orientačně lze anémii rozdělit na mírnou, střední a těžkou, s ohledem na výše zmíněné symptomy a na stupeň omezení fyzické výkonnosti.
!!!začátek petitu!!!
Koncentrace hemoglobinu v krvi jsou nejčastěji 130–160 g/l krve. Zcela orientačně lze pak za mírnou anémii považovat anémii s koncentrací hemoglobinu 110–90 g/l krve, střední s koncentrací hemoglobinu 90–60 g/l krve a těžkou s koncentrací hemoglobinu 60–30 g/l krve. Symptomy spojené s anémií různého stupně však též záleží na příčině a rychlosti, s jakou anémie vznikla, a na eventuálních jiných onemocněních, které mohou současně zhoršovat transport kyslíku (zvláště nemoci respiračního aparátu, které snižují paO2, a nemoci cirkulace, které snižují průtok krve tkáněmi).
!!!konec petitu!!!
Adaptace na anémii spočívá ve zvýšeném srdečním výdeji a v hyperkinetické cirkulaci. Ta je následkem snížení periferního odporu proudění krve cévním řečištěm. Periferní odpor se sníží jednak v důsledku vazodilatace způsobené tkáňovou hypoxií, jednak následkem snížení viskozity krve. Adaptace na anémii dále spočívá ve snížení afinity krve ke kyslíku, tj. posunu disociační křivky kyslíku doprava (v důsledku nadromadění 2,3-DPG v erytrocytech). První druh adaptace je rychlý a uplatňuje se především při fyzické námaze. Je náročný z energetického hlediska – vyžaduje zvýšení srdeční práce. Druhý typ adaptace je energeticky málo náročný, ale vyžaduje pro rozvinutí asi 24 hodin.
Obr.1.11 Znázornění vlivu anémie na parciální tlak kyslíku v různých částech cirkulace. * – vyšší stupeň anémie nebo zvýšení spotřeby kyslíku při anémii
2Řada cytokinů působí multifunkčně, jiné synergizují s jinými faktory.
Tabulka 1.2 Růstové a regulační faktory v krvetvorné tkáni a jejich receptory2
| Zkratka | Růstový faktor | Receptor |
| Funkce: přežívání a stimulace kmenových a časných progenitorových buněk | ||
| SCF | faktor kmenových buněk | c-kit (CD117) |
| IL-3 | interleukin 3 | IL-3R |
| FL | flt-3-ligand | Flt-3 |
| IL-6 | interleukin 6 | IL-6R |
| GM-CSF | granulocyto-makrofágový kolonie stimulující faktor | GM-CSFR |
| IL-1 | interleukin 1 | IL-1R |
| TPO | trombopoetin | mpl |
| Funkce: proliferace a diferenciace pozdních progenitorů | ||
| EPO | erytropoetin | EPOR |
| G-CSF | granulocytový kolonie stimulující faktor | G-CSFR (CD114) |
| M-CSF | monocyto-makrofágový kolonie stimulující faktor | fms (CD115) |
| IL-5 | interleukin 5 | IL-5R |
| IL-7 | interleukin 7 | IL-7R |
| IL-12 | interleukin 12 | IL-12R |
| Funkce: inhibice dělení nebo diferenciace progenitorů | ||
| MIP-1alfa | makrofágový zánětový protein | CKR-1 |
| IL-10 | interleukin 10 | IL-10R |
| TGF-beta | transformující růstový faktor beta | TGF-betaR |
| IFN-gama | interferon gama | IFN-gamaR |
| TNF | faktor nekrotizující nádorové buňky alfa | TNFR |
| Funkce: migrace progenitorových buněk | ||
| SDF-1 | faktor stromálních buněk | CXCR-4 |
| IL-8 | interleukin 8 | CKR-1 (CD128) |
| VEGF | vaskulární endotelový růstový faktor | VEGFR-1,2 |
| Umělé růstové faktory připravené rekombinantními technologiemi | ||
| KW-2228 | marograstim, stabilizovaný G-CSF | G-CSFR (CD114) |
| pIXI-321 | chimerický IL-3/GM-CSF se zvýšenou aktivitou | |
| CH925 | chimerický IL-6/IL-2 | |
| Flt3/G-CSF | chimerický flt-3/G-CSF (progenipoietin) | |
| IL-3/TPO | chimerický IL-3/TPO (promegapoietin) | |
Tabulka 1.3 Některé povrchové antigenní znaky a charakteristiky krevních a progenitorových buněk
| Různé typy progenitorových buněk | CD34, AC133, CD117 (c-kit), nízká exprese CD38 |
| Zralé krevní buňky | |
| panleukocytové znaky | CD45, CD11a/CD18 |
| T-lymfocyty | CD2, CD3, CD4, CD8, CD25 (IL-2Ralfa) |
| B-lymfocyty | CD19, CD20, CD40, Ig |
| plazmatické buňky | CD27, CD38 |
| NK-buňky | CD16 |
| granulocyty | CD15 |
| monocyty | CD4, CD11b, CD14 |
| megakaryocyty | CD61 |
| erytroidní buňky | CD71 (transferinový receptor), glykoforin A |
| dendritické buňky | CD4, CD68, CD80 |
| Antigen prezentující buňky (MHC class II) | HLA-DR, HLA-DQ, HLA-DP |
| Adhezivní molekuly na hematopoetických a stromálních buňkách | CD2 (LFA-2), CD11a, b, c (integriny), CD18 (LFA-1), CD31 (PECAM-1), CD34 |
Obrázek 1.2 Schéma hematopoezy. Kmenové buňky jsou (HSC) přes stadia multipotentních (MPP) a oligopotentních (CMP; CLP, GMP; MEP; EP) progenitorů jsou zdrojem krevních buněk. Sebeobnovná dělení, která nejsou provázená diferenciací , jsou podstatou nevyčerpatelné schopnosti tkáně vytvářet nové funkční buňky. Kmenové buňky lze transplantovat; sebeobnovnými děleními po transplantaci obnoví svoji populaci.
Jedná se o heterogenní skupinu vrozených i získaných onemocnění, které jsou charakterizovány přítomností prstenčitých sideroblastů v krvetvorné tkáni. Sideroblasty jsou erytroidní prekurzory s nahromaděním železa v mitochondriích. Při barvení na železo pruskou modří se toto železo znázorní jako modrý prstenec okolo buněčného jádra, barvitelné železo je obsaženo v mitochondriích. Příjem železa erytroidními prekurzory se zvyšuje v souvislosti s poruchami syntézy hemu. Nepřekvapuje proto, že sideroblastové anémie vznikají při otravě olovem, kdy je inhibován poslední enzym hemové syntézy, ferochelatáza, při podávání protituberkulózního léku izoniazidu a při nedostatku pyridoxinu (vitaminu B6), což jsou stavy, které inhibují hemovou syntézu na samém jejím počátku. Méně jasný je důvod vzniku sideroblastů u některých forem myelodysplastického syndromu typu RARS (refraktory anemia with ring sideroblasts), což je klonální choroba získaná. Hereditární sideroblastová anémie je způsobena snížením syntézy hemu23.
Redukovaná forma kyseliny listové, kyselina tetrahydrolistová (aktivní forma kyseliny listové), se účastní syntézy tymidinu (deoxytymidin-monofosfátu), utilizovatelného pro syntézu DNA18, a purinů, rovněž potřebných pro syntézu DNA, protože jsou součástí nukleotidových purinových bází. Aktivní forma kyseliny listové plní tuto funkci přenosem jednouhlíkových sloučenin („zbytků“). Jejich hlavním zdrojem pro tetrahydrofolát je aminokyselina serin.
Vitamin B12 (kobalamin) je potřebný pro vznik kyseliny tetrahydrolistové z N5-metyltetrahydrofolátu při konverzi homocysteinu na metionin (obr. 1.15). Anémie a jiné hematologické manifestace nedostatku vitaminu B12 a kyseliny listové mají tedy společného jmenovatele, a tím je nedostatek metabolitů kyseliny listové. Velkými dávkami kyseliny listové lze proto odstranit nepříznivé účinky nedostatku vitaminu B12 na krvetvorbu, ale ne naopak.
!!!obr. 15!!!
Přestože účinky nedostatku kyseliny listové a vitaminu B12 na krvetvorbu jsou stejné, projevy onemocnění se mohou lišit. Je to způsobeno především další metabolickou funkcí vitaminu B12, kterou je syntéza metioninu z homocysteinu. Nedostatek vitaminu B12, na rozdíl od nedostatku kyseliny listové, může způsobit poškození CNS, spočívající v demyelinizaci axonů až v zániku neuronů. Hromadění homocysteinu způsobuje hyperhomocysteinémii, která může být příčinou vzniku krevních trombů.
Jak nedostatek kyseliny listové, tak i nedostatek vitaminu B12 způsobují v krvetvorné tkáni poruchu buněčného dělení. Erytropoeza zahrnuje intenzivní buněčné dělení, a je proto k nedostatku těchto dvou vitaminů velmi citlivá. Narušena je však i granulopoéza a tvorba krevních destiček. Nejsou popsány poruchy tvorby lymfocytů. Naproti tomu jsou dobře známy poruchy epitelu sliznic trávicího ústrojí, buněk, které se také normálně intenzivně obměňují za účasti buněčného dělení19.
Anémie, která vzniká v důsledku nedostatku kyseliny listové nebo vitaminu B12, je označována jako megaloblastická a makrocytární, protože buňky v kostní dřeni i erytrocyty v krvi mají větší objem než obvykle. Jejich cytoplazma „dozrává“ (v případě erytroblastů se hemoglobinizuje) dobře, avšak doba mezi buněčnými děleními je významně prodloužená. Objem buňky se proto dlouho zvětšuje, než dojde k jeho redukci buněčným dělením. Prekurzory krevních buněk setrvávají v kostní dřeni déle, a proto je její celularita normální nebo zvýšená. V případě erytroblastů je porucha jejich maturace často (uvádí se, že až v 90 %) provázena jejich zánikem v krvetvorné tkáni, před dozráním do stadia retikulocytu. Tento patologický jev se označuje jako neefektivní erytropoeza. Její součástí je hemolýza již hemoglobinizovaných buněk. Jejím projevem proto může být zvýšená hladina nekonjugovanégo bilirubinu a izoenzymu 1 (erytrocytového) laktát-dehydrogenázy. Přestože může být kostní dřeň hypercelulární, v krvi je velmi málo retikulocytů. Stav kostní dřeně a výskyt retikulocytů v krvi se mohou velmi rychle změnit po podání vitaminu B12 nebo kyseliny listové. Tato dramatická změna, ke které dochází během 2–3 dnů, bývala využívána k potvrzení diagnózy. Dnes lze zjistit případný nedostatek některého nebo obou těchto vitaminů změřením jejich koncentrace v plazmě (norm. 200–900 ng/l vitaminu B12 a 6–20 mg/l kyseliny listové).
Porušená maturace granulocytů a megakaryocytů může způsobovat jistý stupeň neutropenie a trombocytopenie. Dominujícím příznakem je však anémie. U granulocytů způsobuje prodloužená maturace hypersegmentaci jejich jádra. V krvi se také mohou vyskytnout ojedinělé erytroidní a myeloidní prekurzory, ojedinělý erytroblast a myelocyt.
Stupeň anémie z nedostatku kyseliny listové nebo vitaminu B12 může být značný. Hematokrit se může snížit až na 0,15. Anémie však vzniká pomalu a adaptace na ni je proto většinou dobře vyvinuta, protože v erytrocytech je zvýšená koncentrace 2,3-DPG.
Protože se primární příčiny a v jisté části i patogeneze anémie z nedostatku kyseliny listové a vitaminu B12 liší, bude dále stručně pojednáno o obou stavech odděleně.
21Atrofie postihuje převážně fundus a korpus žaludku, méně část prepylorickou. Chybí proto také sekrece kyseliny chlorovodíkové a pepsinu, hladina gastrinu bývá zvýšena. Sekrece kyselé žaludeční šťávy nelze dosáhnout podáním pentagastrinu. Tento stav se označuje jako pentagastrin-rezistentní achlorhydrie. Zvyšuje riziko vzniku karcinomu žaludku.
Krvetvorná (hematopoetická) tkáň se u dospělého člověka nachází v kostní dřeni a v lymfatických orgánech, tj. ve slezině, tymu a v lymfatických uzlinách, ale také v lymfatické tkáni asociované převážně s respiračním a gastrointestinálním orgánovým systémem (označované zkratkou MALT – mucosa associated lymphatic tissue). Krvetvornou tkáň tvoří jednak stroma – buňky a extracelulární část stromatu, a jednak vlastní krvetvorné buňky.
Krvetvorba (hematopoéza)je proces, při kterém dochází k produkci zralých krevních buněk (Obr. 1.2). Ty vznikají postupnou diferenciací z hematopoetických kmenových buněk (HSC, hematopoietic stem cell), přes stadia hematopoetických progenitorů a prekurzorů (viz dále) až do stadia erytrocytů, granulocytů, monocytů, žírných buněk, lymfocytů, dendritických buněk, trombocytů.
!!!obr. 1.2.!!!
Denní potřeba je asi 2,5 μg, přičemž zásoba tohoto vitaminu je asi 4000 μg. Nově vzniklá negativní bilance
mezi jeho příjmem a spotřebou se proto normálně projeví až za několik let. Vitamin B12 je obsažen jen v mase a v mléčných a vaječných produktech. Nedostatek z nedostatečného příjmu potravou proto hrozí pouze u veganů. Hlavními důvody nedostatku vitaminu B12 proto je jeho nedostatečná resorpce gastrointestinálním traktem.
Resorpce vitaminu B12 zahrnuje jeho uvolnění z potravy během žaludeční fáze digesce a vazbu na haptocorin (vazebný protein R, R-faktor). Z této vazby je vitamin B12 uvolněn v duodenu a zde se naváže na vnitřní faktor, glykoprotein secernovaný parietálními buňkami především ve fundu žaludku. Tento komplex je odolný vůči střevní digesci, a dostává se proto až do dolního ilea, kde enterocyty obsahují specifický receptor pro tento komplex, který je celý internalizován do enterocytů. V enterocytech je vitamin B12 uvolněn a navázán na nosič, transkobalamin II. V této podobě se dostává do plazmy. Tento komplex je intenzivně vychytáván hepatocyty, buňkami kostní dřeně a jinými buňkami. Vnitřní faktor je v enterocytech degradován.
Chybění vnitřního faktoru nebo existence protilátek proti vnitřnímu faktoru jsou důležité příčiny vzniku nedostatku vitaminu B12. Chybění vnitřního faktoru způsobí pravidelně totální gastrektomie, méně často částečná gastrektomie. Jinou příčinou je atrofie žaludeční sliznice při atrofické gastritidě. Atrofická gastritida může být způsobena imunitním (autoimunitním) mechanismem – protilátkami proti parietálním buňkám žaludeční sliznice (jejich poškození vazbou a aktivací komplementem), případně protilátkami proti vnitřnímu faktoru. Megaloblastická, makrocyt´ární anémie, která se vyvine následkem atrofické gastritidy vzniklné na autoimunitním podkladě21, se označuje jako perniciózní anémie22.
Jinou příčinou nedostatku vitaminu B12 může být resekce dolního ilea nebo chronické záněty v této části tenkého střeva (regionální enteritida např. u Crohnovy choroby, Whippleova nemoc, tuberkulóza). Snížené vstřebávání vitaminu B12 také způsobuje tropická sprue. Anatomické abnormality v tenkém střevě (divertikuly, anastomózy, pooperační „slepé kličky“, striktury, Crohnova choroba, amyloidóza aj.) mohou způsobit jeho dlouhodobou masivní bakteriální kolonizaci (normálně obsahuje tenké střevo minimum bakterií díky stálému „čištění“ peristaltickými pohyby). To může být také spojeno s nadměrnou spotřebou vitaminu B12 bakteriemi.
Nedostatek vitaminu B12 se může projevovat, vedle hematologické manifestace, rozličnými neurologickými projevy: parestéziemi, zeslabenými nebo zesílenými reflexy, poruchami hlubokého čití a motoriky, poruchami udržování rovnováhy, poruchami kognitivních funkcí až psychotickým stavem. Tyto poruchy jsou způsobeny demyelinizací axonů a následným zánikem míšních i mozkových neuronů. Demyelinizace je přičítána poruše přeměny homocysteinu na metionin způsobené nedostatkem vitaminu B12. Metionin je potřebný pro tvorbu cholinu a fosfolipidů obsahujících cholin. Uvádí se, že toto poškození nervového systému může vzniknout, aniž by se nezbytně projevila porucha krvetvorby. Důležité je to, že poškození nervového systému, na rozdíl od poruchy krvetvorby, může být trvalé. Dodání vitaminu B12 pak odstraní poruchu krvetvorné tkáně, ale ne tkáně nervové.
23Někdy je léčitelná pyridoxinem, který je potřebný pro syntézu hemu.
22Dříve to bývalo onemocnění smrtelné (perniciózní = zhoubný, zničující). Vžitý termín se stále užívá, přestože se prognóza tohoto onemocnění výrazně zlepšila.
Krvetvorba a její poruchy zahrnují procesy, jakými jsou buněčná proliferace, diferenciace, funkční vyzrávání buněk, migrace a apoptóza buněk, genetické změny ap. Jsou to procesy biologické a molekulární povahy, pro jejichž popis je používáno hodně zkratek. Také moderní hematologie proto používá řadu zkratek. Některé tyto zkratky jsou pro ilustraci uvedeny v tabulkách 1.2 a 1.3.
!!!tab. 1.2.–1.3.!!!
18Nedostatek tymidinu se projevuje inkorporací uracilu do DNA. Vzniká patologická DNA bohatá na uridin. Předpokládá se, že nedostatek kyseliny listové v časném těhotenství může způsobit vývojové poruchy spočívající v neuzavření kaudální části páteře (tzv. spina bifida).
19Tyto změny jsou pozorovatelné na sliznici jazyka, mohou způsobovat ragády ústních koutků, poruchy resorpce ve střevě (včetně resorpce kyseliny listové, event. vitaminu B12), průjem.
!!!začátek petitu!!!
Tento stav, charakterizovaný megaloblastovým obrazem v kostní dřeni, může vzniknout poměrně náhle (během několika dnů) buď u nemocných vyžadujících intenzivní lékařskou péči spojenou s hemodialýzou, parenterální výživou a opakovanými krevními transfúzemi, nebo po narkóze s použitím oxidu dusného („rajský plyn“). Příčinou je akutní nedostatek vitaminu B12 nebo kyseliny listové, protože stav se rychle upraví po podání těchto dvou vitaminů. Mechanismus, který způsobí aktutní nedostatek těchto vitaminů, není znám.
!!!konec petitu!!!
!!!začátek petitu!!!
Syntézu DNA narušují některé protinádorové léky. Jejich podávání může způsobit v kostní dřeni podobné změny jako nedostatek kyseliny listové. Jedná se o analogy purinových (např. 6-merkaptopurin, 6-tioguanin, azatioprin) a pyrimidinových (např. 5-fluorouracil a cytozin-arabinozid) nukleotidových bází a některá jiná cytostatika (hydroxyurea, prokarbazin). Rovněž protivirový lék na bázi pyrimidinového analoga, AZT (3´-azido-2´,3´-dideoxytymidin, též zidovudin), používaný k léčbě infekce HIV, často způsobuje megaloblastickou krvetvorbu a anémii.
!!!konec petitu!!!
Tabulka 1.1 Odhad denní produkce „myeloidních“ krevních buněk u dospělého člověka
| Počet buněk/litr | Celkový počet buněk v krvi(5 litrů) | Délka života (Ery)nebo poločas setrvání v cirkulaci(T1/2) (dny) | Počet buněk vytvořenýchběhem 24 hodin(x 108) | |
| Erytrocyty (Ery) | 4,5 x 1012 | 2,25 x 1013 | 120 | 1875* |
| Leukocyty | 6,0 x 109 | |||
| neutrofilní granulocyty65 %** | 3,9 x 109 | 2,0 x 1010 | 0,3 | 462 |
| eozinofilní granulocyty(3 %)** | 1,8 x 108 | 9,0 x 108 | 0,3 | 21 |
| bazofilní granulocyty0,5 %** | 1,5 | 9,0 x 108 | 0,3 | 3 |
| monocyty4 %** | 2,4 x 108 | 1,2 x 108 | 0,3 | 28 |
| (Lymfocyty 27,5 %***) | ||||
| Trombocyty | 1,5 x 1011 | 1,5 x 1012 | 4,5 | (2310) |
| „megakaryocyty“**** | 1 |
Anémii může způsobit krvácení akutní nebo krvácení chronické. Pro patofyziologii anémie způsobené akutní ztrátou krve je podstatné množství krve, které unikne z cévního systému v důsledku porušení jeho uzavřenosti při úrazech (také při krvácení do abdominální dutiny), během operací či v souvislosti s porodem nebo u dárců krve. Ztráta 50 ml krve představuje ztrátu jen asi 1 % a ztráta 500 ml jen asi 10 % erytrocytů. Je zřejmé, že jen větší krvácení, nad 500 ml krve, způsobí významný stupeň anémie. Anémie, definovaná jako snížení koncentrace hemoglobinu, počtu erytrocytů v jednotkovém objemu krve, jakož i snížení hodnoty hematokritu, se vyvine během jedné hodiny nebo několika málo hodin v důsledku regulačních pochodů zaměřených na udržení velikosti cirkulujícího objemu krve.
!!!začátek petitu!!!
Narušením Starlingovy kapilární rovnováhy nízkým intravaskulárním tlakem dojde k přesunu části intersticiální tekutiny do plazmy a současně volumoregulační mechanismy, ovlivňující konečný objem moče, působí ve smyslu zadržení potřebného objemu vody a iontů k doplnění objemu tělesných tekutin, včetně celkového objemu krve. V důsledku těchto dějů se zvětšuje objem plazmy, a proto klesá hematokrit.
!!!konec petitu!!!
Ztráta erytrocytů obsažených v 1000 ml krve je spojena se ztrátou asi 500 mg železa. To je množství, které může u ženy v reprodukčním období představovat více, než jsou celkové zásoby železa v těle. U muže se toto množství může přiblížit celkové zásobě železa.
Pokles koncentrace hemoglobinu po akutním krvácení stimuluje tvorbu erytropoetinu, jehož koncentrace se zvýší během několika hodin úměrně stupni anémie. Se zpožděním čtyř až pěti dnů po akutním krvácení se může v krvi významně zvýšit výskyt retikulocytů a anémie se začne upravovat. Nedostatek železa v zásobárnách může tento pochod zpomalit, protože získání železa jeho zvýšenou resorpcí z potravy vyžaduje delší čas.
V patofyziologii chronického krvácení, v rozsahu desítek ml krve za den, hraje významnou roli kumulovaná ztráta železa, která může převýšit možnosti jeho doplňování zvýšenou resorpcí z potravy. Vyčerpání zásob železa v tomto případě omezuje kompenzační možnosti erytropoezy. Po krvácení se tvorba erytrocytů zvýší méně než v případě rozpadu erytrocytů hemolýzou, kdy železo zůstává v organismu. Následkem dlouhodobého chronického krvácení může dojít k vyčerpání zásobáren železa, kompenzační možnosti krvetvorné tkáně tím mohou být významně omezeny a krevní obraz může jevit znaky anémie z nedostatku železa.
Obr. 1 V embryogenezi vzniká cévní systém a krev společně a paralelně. Zajištění stálé a intenzivní komunikace mezi tkáněmi a orgány celého organismu nemohou jeden bez druhého zajistit.
Obr. 1.16 Schéma buněčné membrány erytrocytů s přichycením spektrinu k lipidové dvojvrstvě
Krev umožňuje stálou a intenzivní komunikaci mezi všemi tkáněmi a orgány organismu. Tuto její funkci umožňuje právě tekutost krve a další vlastnosti, které se souhrnně označují jako reologické vlastnosti krve. Při výčtu funkcí krve je proto třeba uvést schopnost krve setrvat v tekutém stavu a udržet si vhodné reologické vlastnosti. Krev je v neustálém kontaktu s endotelovou výstelkou cévního systému, jejíž plocha je odhadována na 1000 m2. Krev a endotelová výstelka cév se vzájemně významně ovlivňují, a často proto jejich funkce nemůžeme posuzovat odděleně (Obr. 1.1).
!!!obr.1.1!!!
Narušení celistvosti cévního systému by vedlo, vzhledem k tekutosti krve, ke ztrátě tohoto orgánu. Důležitou funkcí krve je proto i její schopnost rychlého přechodu z tekutého stavu ve stav pevný, který napodobuje konzistenci jiných orgánů. Další důležitou funkcí krve je pak její schopnost zpětného obnovení své tekutosti, tj. změny z „pevného“ stavu ve stav tekutý.
Transportní funkce krevní plazmy (dále jen plazma) je zesílena přítomností transportních molekul, které umožňují přenos specifických látek jiným způsobem než jejich prostým rozpouštěním. Tento transport se týká řady velmi různorodých látek, např. železa, mědi, steroidních a tyreoideálních hormonů, hemoglobinu a heminu uvolněných z erytrocytů, lipidů aj.
V krvi jsou přítomny specifické látky, které jsou zdrojem informací pro buňky vybavené příslušnými receptory – jedná se o klasické hormony, růstové faktory, cytokiny a jejich inhibitory.
Schopnost určitých složek krve opouštět cévní systém a opět se do něho vracet je další významnou funkcí krve. Například stálá výměna vody a iontů mezi plazmou a intersticiální tekutinou je velmi intenzivní. Ve specifickém případu glomerulární filtrace v ledvinách se jedná asi o 180 litrů/den.
Významná funkce krve spočívá i v její účasti na imunitních reakcích, a to jednak díky přítomnosti různých typů imunitních buněk účinných jak v oblasti přirozené, tak i získané imunity, a jednak díky přítomnosti specifických imunoglobulinů. Součástí krvetvorného systému jsou proto i periferní lymfatické tkáně ve slezině, asi 500 lymfatických uzlinách a v thymu.
Krev se, vedle lokálních faktorů, účinně podílí na rozvoji a průběhu zánětlivé reakce a při hojení ran. To jsou důležité fyziologické mechanismy sloužící k udržení integrity tkání organismu při jeho interakci se zevními patogenními podněty.
Další významnou funkcí krve je přenos kyslíku a oxidu uhličitého. Jde o dodání asi 250–3500 ml kyslíku za minutu tkáním, v závislosti na aktuální tělesné aktivitě, a o transport asi o 20 % menšího objemu oxidu uhličitého (při respiračním kvocientu 0,8) z tkání do plic. Pro tyto funkce je významná přítomnost erytrocytů a hemoglobinu v nich obsaženém. V případě kyslíku, který se při teplotě 37 °C ve vodě velmi málo rozpouští, zvyšuje přítomnost hemoglobinu jeho obsah v krvi asi 50krát. V případě oxidu uhličitého, který má vyšší koeficient rozpustnosti ve vodě a je přenášen také ve formě bikarbonátu, je význam hemoglobinu při jeho přenosu menší, avšak též významý.
Krev má významnou úlohu v udržování acidobazické rovnováhy ve vnitřním prostředí organismu.
Různé komponenty krve jsou obměňovány s různou intenzitou. Týká se to jak plazmatických bílkovin, jejichž hlavním zdrojem jsou játra, tak i jaderných krevních buněk a nejaderných krevních elementů, erytrocytů a trombocytů (dále jen „krevních buněk“). Jejich zdrojem je krvetvorná tkáň. Krvetvorná tkáň je orgán o celkové hmotnosti asi 2,5 kg u dospělého jedince. Vzhledem k významu krvetvorné tkáně pro obměnu krevních buněk představují krev a krvetvorná tkáň integrální orgánový systém. Vedle kostní dřeně patří k tomuto orgánovému systému ještě slezina, tymus a lymfatická tkáň1. Hlavní funkcí krvetvorné tkáně je stálá produkce různých druhů krevních buněk a krevních elementů, a to v potřebném množství a správném stupni funkční vyzrálosti. Přibližné množství jednotlivých druhů krevních buněk a krevních elementů produkovaných krvetvornou tkání dospělého člověka je uvedeno v tabulce 1.1. Je zřejmé, že kostní dřeň produkuje převážně erytrocyty a neutrofilní granulocyty, pokud trombocyty považujeme za „úlomky“ megakaryocytů a megakaryocyty za buňky vytvářené kostní dření.
!!!tab. 1.1.!!!
Hereditární sférocytóza (HS) je ve většině případů (75%) autozomálně dominantně dědičné onemocnění, které je způsobené defektem cytoskeletu a buněčné membrány. Pro HS je typické snížené množství spektrinu v cytoskeletu, které bývá způsobeno defektem některého z proteinů, které zprostředkovávají jeho ukotvení k buněčné membráně erytrocytu (ankyrin, protein pásu 3, protein 4.2), spíše než defektem spektrinu samotného (Obr 1.16).
!!!začátek petitu!!!
Hereditární eliptocytóza je naopak nejčastěji způsobena mutacemi genů kódujích α nebo β spektrin s výslednou abnormální tvorbou spektrinových heterodimerů.
!!!konec petitu!!!
!!!obr. 1.16!!!
Lipidová dvojvrstva erytrocytové membrány je nedostatečně zakotvena ke spektrinové vrstvě a dochází v ní k tvorbě vezikul. Při průchodu sinusoidy sleziny dochází ke ztrátám části erytrocytové membrány. Povrch erytrocytu se zmenšuje v poměru k jeho vnitřnímu objemu. Erytrocyt ztrácí svůj charakteristický bikonkávní tvar, stává se kulovitým, sférickým nebo elipsoidním, případně nabývá jiného charakteristického tvaru (stomatocytů). Vždy se snižuje jeho deformabilita a odolnost k opakovaným průchodům kapilárami, zvláště stěnou slezinných sinusů. Z tohoto důvodu má splenektomie většinou výrazný léčebný efekt, který významně prodlužuje setrvání sférocytů v cirkulaci a snižuje rozsah jejich hemolýzy. Pro krevní obraz je samozřejmě nadále příznačná přítomnost patologických forem erytrocytů.
V klinickém obraze dominuje, vedle anémie, která může být z důvodu kompenzace mírná, splenomegalie, ikterus, event. komplikace způsobené žlučovými kameny. Pro onemocnění je charakteristická přítomnost malých sférocytů v krevním nátěru při jen mírně sníženém průměrném objemu erytrocytů (MCV) a zvýšený počet retikulocytů v krvi.
!!!začátek petitu!!!
Nesrážlivá krev, inkubovaná 48 hodin při teplotě 37 °C vykazuje normálně jen nízký stupeň hemolýzy erytrocytů (< 4 %). Při sférocytóze nebo eliptocytóze to může být 10–50 %. Sférocyty jsou méně rezistentní vůči hypotonickému prostředí zředěného fyziologického roztoku. Normální erytrocyty začínají hemolyzovat až při snížení koncentrace NaCl pod 5–4 g/l, kdežto erytrocyty od nemocného se sférocytovou anémií již při koncentraci 7–6 g/l24.
!!!konec petitu!!!
U některých jedinců, častěji u mužů (obézních, kuřáků, se zvýšeným arteriálním krevním tlakem) mohou být hodnoty červeného krevního obrazu zvýšené s hodnotou hematokritu okolo 0,55. Je to způsobeno zmenšením celkového krevního objemu, jdoucím plně na vrub jeho plazmatické části.
!!!začátek petitu!!!
Radioizotopové vyšetření celkového množství erytrocytů prokázala v těchto případech jejich normální množství. Normální je i aktivita erytropoezy v krvetvorné tkáni a výskyt retikulocytů v krvi, ani hladina erytropoetinu není zvýšena.
!!!konec petitu!!!
Jedná se tedy o poruchu volumoregulace krevního (plazmatického) objemu, ne o poruchu krvetvorné tkáně. Příčina není známa. Zvýšená viskozita krve zvyšuje riziko vzniku trombů, může se podílet i na patogenezi arteriální hypertenze.
24Koncentrace NaCl ve fyziologickém roztoku je 9 g/l.
Vedle pobytu ve vysokohorském prostředí mohou tkáňovou hypoxii způsobit onemocnění respiračního aparátu a ty srdeční a cirkulační vady, které způsobují příměs venózní krve ke krvi arteriální (pravo-levý zkrat).
Paroxyzmální noční hemoglobinurie (PNH) je vzácné získané onemocnění krvetvorby, nádorového benigního charakteru způsobeného krvetvorbou z mutované kmenové buňky, které se projevuje: hemolytickou anémií - hemolýza je intravaskulární a je zprostředkovaná aktivací komplementu, zvýšeným rizikem vzniku trombóz (venózních i arteriálních), rizikem vzniku selhání kostní dřeně (vznikem aplastické anémie). PNH vzniká v důsledku mutace genu PIG-A. Gen PIG-A kóduje enzym nezbytný pro vytvoření tzv. fosfatidyl-inozitol-glykozylové (PIG) kotvy, kterou jsou k buněčné membráně ukotveny některé proteiny. Inaktivační mutace genu PIG-A způsobí, že na povrchu buněk patologického klonu chybí skupina proteinů (je jich dnes známo více než 20), které nejsou částí buněčné membrány, nýbrž jsou k ní vázány výše zmíněnou GIT molekulární kotvou. Mezi tyto proteiny patří i dva proteiny (MIRL a DAF), jejichž funkcí je ochrana buněčné membrány vůči lytickým účinkům komplementu. Nejčastěji se označují podle monoklonálních protilátek, které s nimi specificky reagují, jako CD55 a CD59. Inaktivují některé složky komplementové kaskády a snižují tak vytváření lytického kompexu (C6–9). V krvi a v kostní dřeni nemocných s PNH jsou přítomny jak normální erytrocyty, tak erytrocyty defektní v CD55 a CD59.
!!!začátek petitu!!!
Deficit těchto proteinů je nejen na provrchu erytrocytů, ale také granulocytů, trombocytů, u kterých však se membránový defekt významně funkčně neprojevuje, až na zvýšené riziko vzniku trombóz. V krvetvorné tkáni jsou přítomny normální klony hematopoetických buněk vedle klonu patologického.
!!!konec petitu!!!
Nejčastějším projevem PNH je chronická hemolytická anémie různého stupně, která bývá normochromní normocytární. Noční záchvaty (paroxyzmy) hemolýzy s ranní hemoglobinurií a tmavě zbarvenou močí jsou jen u části pacientů. Ataky hemolýzy během noci s následnou ranní hemoglobinurií se vysvětlují mírným okyselením vnitřního prostředí organismu během spánku. Klinicky nejzávažnější komplikací je vznik trombózy v různých lokalizacích, arteriální či venózní. Nejčastější jsou trombózy intraabdominální, trombózy hepatálních žil (tzv. Budd Chiariho syndrom) a trombózy mozkových žilních splavů. Mechanismus zvýšené náchylnosti ke vzniku trombóz není jasný.
!!!začátek petitu!!!
K diagnostice se v minulosti používal Hamův test. Ten spočívá v inkubaci erytrocytů v normálním séru okyseleném na pH 6,2. Kyselé protředí aktivuje komplement alternativní cestou a patologické erytrocyty podlehnou hemolýze. K potvrzení diagnózy PNH se dnes používá průtoková cytometrie s průkazem deficitu proteinů CD55 a CD59 na povrchu erytrocytů či na povrchu granulocytů nebo monocytů. V laboratorních nálezech jsou dále známky hemolýzy (zvýšený počet retikulocytů, zvýšení LDH, snížený/spotřebovaný haptoglobin, hemoglobinurie a hemosiderinurie).
Hemoglobinurie je spojena se ztrátami železa močí, kterou se normálně prakticky žádné železo z těla neztrácí. Na druhé straně, pokud silné a pravidelné ataky hemolýzy vyžadují dlouhodobou léčbu podáváním erytromasy, může se železo v těle hromadit.
Pacienti s PNH mohou mít v krevním obraze nejen anémii, ale také leukopenii a trombocytopenii různého stupně.
!!!konec petitu!!!
25Záměna lyzinu za glutamovou kyselinu v 6. pozici β-řetězce.
1Řada cytokinů působí multifunkčně, jiné synergizují s jinými faktory.
Srpkovitá anémie (sickle cell anemia) je onemocnění způsobené záměnou jedné aminokyseliny v β-globinovém řetězci (místo hydrofilní kyseliny glutamové je na 6. pozici přítomen hydrofobní valin). Toto je dáno bodovou mutací β-globinového genu v 6. kodónu (substitucí 1 nukleotidu, GAG 🡪 GTG). Hemoglobin s touto aminokyselinovou záměnou bývá označován písmenem S (SS u homozygota, AS u heterozygota). Termín srpkovitá anémie je dnes používán převážně pro homozygotní stav, kdy jsou oba řetězce β postiženy touto charakteristickou mutací.
!!!začátek petitu!!!
Podobný těžký klinický obraz má i kombinace výskytu hemoglobinu S s talasémií β0 nebo β+ (viz dále) nebo s hemoglobinem C (genotyp SC)25.
!!!konec petitu!!!
Heterozygotní stav hemoglobinu AS má jen velmi malé klinické projevy. Vyskytuje se však až s 8% frekvencí u černošské populace v USA a Afričanů v některých částech Afriky.
Hemoglobin S mění své fyzikální vlastnosti, je-li v deoxygenované formě. Hydrofobní zbytek valinu se „zanořuje“ do hydrofobní oblasti sousedních β-řetězců. Snižuje se rozpustnost hemoglobinu a molekuly hemoglobinu vzájemně polymerizují, což způsobuje snížení deformability erytrocytů a charakteristickou změnu jejich tvaru, srpkovatění. Erytrocyty proto cykliciky mění tvar při průchodu kapilárami, protože zde je z hemoglobinu odebírána část kyslíku. Při průchodu plícemi a nasycení kyslíkem se jim vrací normální bikonkávní tvar. Protože k těmto změnám tvaru dochází opakovaně, více než tisíckrát za den, patologický srpkovitý tvar se po určité době fixuje (ireverzibilně srpkovité erytrocyty). Hemoglobin v erytrocytech také částečně denaturuje a dochází i k poškození erytrocytové membrány. Na povrch erytrocytů se zvýšeně váží imunoglobuliny IgG, což je senzibilizuje vůči zachycení a destrukci makrofágy. Klinické projevy srpkovité anémie jsou dány zejména vznikem vazo-okluzivních krizí a přítomností chronické hemolytické anémie, která bývá středně těžká až těžká. Životnost erytrocytů je zkrácena z normální 120 dnů na 10-25 dnů. Anémie může být i dobře tolerována díky rozvoji kompenzačních mechanismů. Laboratorně jsou vedle anémie přítomny markery hemolýzy. Dále je patrný ikterus na sliznicích a na očním bělmu a bývá tendence ke vzniku žlučových konkrementů. Slezina nebývá zvětšená, naopak může být velmi malá a kalcifikovaná v důsledku opakovaných infarktů způsobených ztíženým průchodem srpkovitých erytrocytů její mikrocirkulací. Může být dokonce zcela nezřetelná (tzv. autosplenektomie).
Snížená průchodnost krve mikrocirkulací a reologické problémy způsobené přítomností srpkovitých erytrocytů jsou příčinou tzv. vazo-okluzivních krizí, při nichž dochází k obstrukcím mikrocirkulace s následnou tkáňovou hypoxií a rozvojem infarktů. Vazo-okluzivní krize se klinicky projevují jako akutně vzniklé bolesti v různých orgánech a tkáních nebo jako náhlé výpadky funkce.
!!!začátek petitu !!!
Náhlé neurologické symptomy zahrnující problémy motorické a senzorické, poruchy zraku z ischemického poškození sítnice, mikroinfarkty plicní projevující se náhlými bolestmi na hrudníku, náhlé intenzívní bolesti svalové, aseptická nekróza hlavy femuru, to vše patří do možné klinické symptomatologie tohoto onemocnění. Mezi faktory, které zvyšují riziko těchto komplikací, patří snížená saturace krve kyslíkem z důvodu vysokohorského pobytu nebo poruše funkce plic, zvýšená extrakce kyslíku z krve protékající mikrocirkulací při zvýšené metabolické aktivitě tkání (např. při cvičení), dehydratace, infekce a stres.
Opakované vazo-okluzivní krize vedou k postupnému poškozování celé řady orgánů. Chronické poškozování orgánů se týká plic s možným rozvojem plicní fibrózy a plicní hypertenze, ledvin (zvláště dřeně ledviny, ve kterých může vzniknout papilární nekróza26 způsobující hematurii), kůže (chronické ulcerace), zraku a funkcí CNS (po opakovaných cévních mozkových přívodách) aj.
!!!konec petitu!!!
Příčinou snížené tvorby granulocytů je nejčastěji iatrogenní poškození kostní dřeně v souvislostí s protinádorovouu léčbou cytostatiky. Jinou příčinou jsou poruchy kostní dřeně spadající do kategorie aplastické anémie a myelofibrózy/myeloftizy. Také nedostatek vitaminu B12 nebo kyseliny listové sníží produkci nejen erytrocytů, nýbrž i granulocytů. Některé bakteriální a virové infekce (tuberkulóza, brucelóza, infekční mononukleóza, hepatitida aj.) mohou být provázeny granulocytopenií, která je též způsobena sníženou tvorbou granulocytů. Existují také vrozené nebo získané formy neutropenie.
!!!začátek petitu!!!
Cyklická neutropenie. Jedná se o velmi vzácné většinou autozomálně dominantně dědičné onemocnění, při kterém se počet granulocytů v krvi přechodně snižuje v cyklech trvajících 19–21 dnů (může dojít až k jejich chybění). Těžká neutropenie trvá několik dnů a v tomto období se zvýšeně vyskytují infekční, převážně bakteriální, onemocnění. V mezidobí jsou počty granulocytů v dolním pásmu normálních hodnot. Onemocnění je způsobeno mutací genu ELANE, který kóduje neutrofilní elastázu. Neutrofilní elastáza je produkována zejména promyelocyty a předpokládá se, že je důležitá pro vývoj neutrofilních granulocytů. Mutace v genu ELANE způsobují akumulaci nefunkčního proteinu v endoplazmatickém retikulu. Akumulace abnormálního proteinu v endoplazmatickém retikulu aktivuje řadu intracelulárních signálních drah a vede k apoptóze. Morfologicky je v kostní dřeni patrná zástava maturace na úrovni promyelocytů. Pacienti s cyklickou neutropenií nemají vyšší riziko vzniku AML.
Kongenitální těžká neutropenie je heterogenní skupinou velmi vzácných onemocnění (patří sem i Kostmannův syndrom), které se vyznačují těžkou vrozenou neutropenií (<0.2 x 109/L). Onemocnění se projeví brzy po narození častými a život ohrožujícími infekcemi. Dědičnost může být autozomálně dominantní, autozomálně recesivní nebo X- vázaná. Významná část případů vzniká sporadicky. Více než polovina onemocnění, které vykazují autozomálně dominantní dědičnost, je způsobená mutacemi genu ELANE, který kóduje neutrofilní elastázu (viz výše). Mutace se však nacházejí v jiné oblasti genu než ty, které způsobují cyklickou neutropenii. V kostní dřeni je porušeno vyzrávání granulocytů ve stadiu promyelocytu a myelocytu. U poměrně vysokého procenta postižených se později vyvine akutní myeloidní leukémie nebo nyelodysplastický syndromu. Tento syndrom je proto považován za „preleukémii“.
Idiopatická neutropenie. Příčina dlouhodobé neutropenie není v případě idiopatické neutropenie zřejmá.
!!!konec petitu!!!
Tabulka 1.14 uvádí stavy, u kterých je zvýšená koncentrace hemoglobinu (počtu erytrocytů, hematokritu) v krvi a které jsou označované jako polycytémie. Onemocnění polycythaemia vera bylo probráno jako součást myeloproliferativních onemocnění. Ostatní polycytémie jsou nenádorové patologické stavy.
!!!tab. 1.14!!!
Jedná se o velmi vzácné stavy, kdy určitá mutace naruší buď regulaci tvorby erytropoetinu, nebo změní citlivost erytroidních progenitorů k účinkům erytropoetinu. Může se také jednat o hemoglobinopatii, která zvýší afinitu krve ke kyslíku. Přenos kyslíku krví je pak méně efektivní a adaptačně se zvýší koncentrace hemoglobinu v krvi.
26Z důvodu hypertonického prostředí se v procházejících erytrocytech zvyšuje koncentrace hemoglobinu, což spolu s fyziologicky nízkým tlakem kyslíku ve dřeni ledviny zesiluje proces srpkovatění.
Polycytémie, při které jsou selektivně zvýšené jen hodnoty červeného krevního obrazu, současně je dostatečná saturace arteriální krve kyslíkem (je normální paO2) a je zvýšená hladina erytropoetinu, je důvodem k vyšetřením zaměřeným na zjištění případného nádorového onemocnění, které by bylo zdrojem ektopické produkce erytropoetinu. Jen velmi malé procento nádorů ledvin, jater, dělohy, mozečku, ev. jiných, produkuje erytropoetin.
Krev představuje „tekutý orgán“ o celkové hmotnosti 5–6 kg u dospělého člověka. Obsahuje část buněčnou a část plazmatickou v objemovém poměru přibližně 0,45 : 0,55. Z fyzikálního hlediska představuje ne-newtonovskou tekutinu, která čím rychleji proudí, tím nižší má viskozitu. Podstata krevních onemocnění je často odrazem poruch funkce krvetvorných tkání.
Ke zvýšeným ztrátám erytrocytů může docházet z důvodu krvácení nebo zkráceného setrvání erytrocytů v cirkulaci v důsledku jejich předčasné hemolýzy.
Jako hemoglobinopatie označujeme stavy, u kterých je změněno aminokyselinové složení některého globinového řetězce nebo se bílkovinné podjednotky hemoglobinu nevytvoří ve správném poměru (talasémie). Bylo popsáno velké spektrum změn v aminokyselinovém složení globinových řetězců, často vykazujících hereditu a velmi vzácný výskyt. Jen část z nich způsobuje hemolytickou anémii, další způsobují změny v afinitě erytrocytů ke kyslíku a následnou kompenzační polycytémii nebo anémii, jiné jsou zcela asymptomatické.
!!!začátek petitu!!!
Jinou hemoglobinopatií způsobující hemolytickou anémii je skupina tzv. nestabilních hemoglobinů, které mají tedenci k precipitaci. Příčinou je většinou mutace, která snižuje pevnost vazby hemu ke globinovým řetězcům nebo snižuje vzájemnou vazbu globinových řetězců v tetrameru hemoglobinové molekuly. Precipitovaný hemoglobin se váže k erytrocytové membráně v podobě tzv. Heinzových tělísek. Jejich přítomnost v krvi je zvláště patrná u splenektomovaných jedinců s nestabilním typem hemoglobinu, protože slezina Heinzova tělíska z erytrocytů odstraňuje.
!!!konec petitu!!!
Příčinou snížení počtu neutrofilních granulocytů v krvi může být jejich nedostatečná produkce, jejich sekvestrace ve zvětšené slezině nebo extravaskulárně a dále jejich zvýšený zánik buď v krvi, nebo po přechodu to tkání.
Granulocytopenii může působit hypersplenismus, při kterém je část krevních granulocytů sekvestrována ve zvětšené slezině. Jinou příčinou přechodné granulocytopenie může být rozsáhlá infekce nebo zánět, kdy se velký počet granulocytů přesune z krve do postižené tkáně.
Tab. 1.14 Patofyziologická klasifikace polycytémií
Polycytémie ze zvýšené stimulace erytropoetinem
Polycytémie v důsledku celulární poruchy krvetvorné tkáně
Tvorba erytropoetinu je zvýšena buď z důvodu tkáňové hypoxie, nebo je zdrojem erytropoetinu nádor. Společným znakem je zvýšená hladina erytropoetinu v plazmě při zvýšené koncentraci hemoglobinu v krvi (viz obr. 1.13).
V těchto případech se většinou jedná o vrozené, často dědičné defekty erytrocytů. Porucha erytrocytů může narušovat funkci buněčné membrány a cytoskeletu, hemoglobinu nebo metabolismu erytrocytu.
Tyto obrázky ze staré učebnice nejsou v novém teextu využity.
Jsou ilustarivní a samovypovídající.
Stojí za zvážením zda by se neměly do nového textu na odpovídajících místech využít.
Talasémie jsou vrozená, většinou dědičná onemocnění, při kterých dochází v erytroidních buňkách k dysbalanci v množství vytvářených α- a β-globinových řetězců (Tab. 1.13). Řetězec α je v genomu kódován čtyřmi alelami (chromozom 16), řetezec β dvěma alelami (chromozom 11).
!!!tab. 1.13.!!!
Alfa-talasémie jsou způsobeny mutacemi – nejčastěji delecemi v oblasti α globinových genů, které vedou k nedostatku α-globinových řetězců a tento nedostatek může mít čtyři stupně závažnosti v závislosti na počtu inaktivovaných alel.
!!!začátek petitu!!!
Chybění jedné čtvrtiny α-řetezců nezpůsobuje žádný patologický fenotyp. Při chybění poloviny α-řetezců (v důsledku inaktivace dvou alel) vzniká jen malá porucha, která se projevuje jen velmi mírnou anémií, s mírnou mikrocytózou a hypochromií erytrocytů. Inaktivace tří alel je příčinou hemolytické anémie, protože nadbytek β-řetězců je již natolik velký, že tyto spolu vytvářejí tetramery, označované jako hemoglobin H (β4). Je to nestabilní hemoglobin, který precipituje a adheruje k cytoskeletu a k membráně erytrocytu za tvorby Heinzových tělísek. Anémie je mikrocytová a hypochromní, s terčovitou formou erytrocytů. Počet retikulocytů je zvýšený a klinický obraz hemolytické anémie s převážně extravaskulární hemolýzou doplňuje splenomegalie. Inaktivace všech čtyř alel pro α-globinové řetězce je neslučitelná se životem. Jediné možné formy hemoglobinu jsou hemoglobin Bartův (4γ) a hemoglobin H (β4), oba nestabilní, s vysokou afinitou ke kyslíku a s hyperbolickou (ne sigmoidní) disociační křivkou kyslíku. Plod buď umírá před narozením (hydrops fetalis), nebo novorozenec umírá krátce po narození.
!!!konec petitu!!!
Beta-talasémie jsou způsobeny mutacemi v oblasti promotoru, kódující sekvenci, rozhraní intronů/exonů nebo v oblasti polyA konce β globinového genu. Mutace postihující jednu alelu β globinového genu lze rozdělit dle jejich dopadu na β+-mutace, při nichž dojde ke snížení produkovaného množství normálních β-řetězců a na β0-mutace, při nichž dochází k úplnému zániku produkce β-řetězců z mutované alely β globinového genu. Různé kombinace těchto patologických alel pak způsobují různou klinickou závažnost onemocnění β-talasémií. Nadbytek α-globinových řetězců poškozuje erytroidní buňky svou vazbou k buněčné membráně. To může způsobovat těžkou hemolytickou anémii, kdy část vytvářených erytroidních buněk se rozpadne již v kostní dřeni. Tento stav se označuje jako neefektivní erytropoeza. Přežívání erytrocytů v cirkulaci může být také velmi podstatně zkráceno, především u homozygotní onemocnění, tzv. thalassaemia major (nebo Cooleyovu anémii). Protože β-řetězce mohou zcela chybět, v krvi je pak místo hemoglobinu A přítomen hemoglobin F (2αγ) a hemoglobin A2 (2αδ).
Intenzivní hemolýza u nemocných s homozygotní formou β-talasémie, trvající od narození, působí intenzívní stimulaci erytropoezy, která kompenzuje velké množství zanikajících erytrocytů. Krvetvorná tkáň je hypertrofická, což způsobuje ztenčení dlouhých kostí a deformity kostí lebky. Časté patologické fraktury dlouhých kostí a jejich následné hojení způsobují růstovou retardaci. Přítomna je také extramedulární hematopoéza v játrech a slezině, což způsobuje hepato/splenomegalii. Erytrocyty mají malý objem (mikrocytóza) a jsou hypochromní. Onemocnění vyžaduje opakované transfúze erytromasy od časného dětství, a to nejen z důvodu udržení dostatečně vysoké koncentrace hemoglobinu, ale i z důvodu snížení celkové stimulace a expanze krvetvorné tkáně. Transfúzemi erytromasy se však do organismu dostává velké množství železa, kterého se organismus nedovede zbavit. Nahromadění železa poškozuje některé orgány pravděpodobně prostřednictvím zvýšené tvorby kyslíkových radikálů. Kritické bývá poškození jater, kde vznikají cirhotické změny. Poškození Langerhansových ostrůvků v pankreatu způsobuje inzulindependentní diabetes mellitus (1. typu). Poškození myokardu je spojeno s fibrotickými změnami, které mohou narušit jeho diastolickou a systolickou funkci a vést k srdečnímu selhání.
Vedle této nejtěžší formy β-talasémie existují formy s čátečně zachovalou produkcí β-řetězců. Intenzita hemolýzy a klinických projevů je úměrně menší. U heterozygotů, se zachovalou produkcí asi 50 % normálního množství β-řetězců, nemusí být anémie vůbec vyjádřena nebo je mírná. Bývá však u nich mikrocytóza s hypochromními erytrocyty a zvýšená koncentrace hemoglobinu A2 (thalassaemia minor).
Tabulka 1.13 Přehled talasémií (pokud je anémie, pak je hypochromní a mikrocytová)
Příčinou může být výskyt protilátek proti neutrofilním granulocytům v rámci autoimunitního onemocnění (autoimunitní neutropenie – AIN). Tvorbu protilátek proti granulocytům může vyvolat příjem některých léků, které se naváží na povrch granulocytů a působí změnu jejich antigenní podoby (působí jako hapten). Tvorba autoprotilátek proti granulocytům může být součástí onemocnění systémovým lupus erythematodes nebo revmatoidní artritidou.
Granulocyty jsou nejčetnějšími typy leukocytů v krvi. Mají důležitou funkci při obraně organismu vůči infekcím, jsou součástí vrození imunity. Snížení jejich počtu v krvi se projevuje častějším výskytem infekcí. Ty jsou často způsobeny stafylokoky nebo streptokoky, ale může se jednat i o infekce jinými bakteriemi, jakož i o infekce způsobené kvasinkami, houbami a parazity. Mezi klinické projevy patří zvýšená teplota, únava, nechutenství, časté bolesti v krku, časté a opakující se ulcerace a záněty v dutině ústní, záněty středního ucha, záněty urogenitální a kožní infekce. Infekce mají protrahovaný a závažný průběh.
!!!začátek petitu!!!
Časté infekce se dostavují, sníží-li se počet neutrofilních granulocytů pod 1 x 109/l krve, a pravidelně se vyskytují při poklesu pod 0,5 x 109/l krve. Při hodnotách neutrofilních granulocytů nižších než 0,2 x 109/l krve je silně potlačena zánětlivá reakce jako obranná reakce na infekci nebo jiné tkáňové poškození.
!!!konec petitu!!!
Vzhledem k velkému obratu neutrofilních granulocytů v krvi, ve které po vyplavení z kostní dřeně setrvávají průměrně jen několik hodin, existenci jejich marginálního poolu v cirkulaci a poměrně velké rezervě v kostní dřeni se může počet granulocytů v krvi poměrně rychle měnit.
Obr.1.11 Vývoj anémie při snížené produkci erytrocytů. A – normální stav; B – snížení produkce erytrocytů pod 2 x 1011/den způsobí zmenšování celkového počtu erytrocytů v krvi, protože jich více zaniká než vzniká; C – po určité době se vytvoří nový rovnovážný stav, ve kterém jsou opět zánik a produkce erytrocytů v rovnováze
Zvýšení počtu granulocytů v krvi může být způsobeno třemi mechanismy: uvolněním granulocytů přichycených k endoteliím především v plicní cirkulaci a ve slezině (působením adrenalinu), vyplavením granulocytů z kostní dřeně do krve (účinkem glukokortikoidů nebo růstového faktoru G-CSF) a konečně zvýšenou produkcí granulocytů kostní dření pod vlivem růstových faktorů G-CSF a GM-CSF. Zvýšená produkce granulocytů a jejich intenzivnější vyplavování ze dřeně do krve bývá provázeno vyšším zastoupením méně vyzrálých forem granulocytů v krvi, forem, které se vyznačují nižším počtem jaderných segmentů nebo protaženým nesegmentovaným jádrem (tzv. tyčky). Hovoří se o „posunu doleva“ v průměrné vyzrálosti cirkulujících granulocytů. Tento nález má podobný význam jako zvýšení počtu retikulocytů. Ukazuje na stimulaci kostní dřeně růstovým faktorem G-CSF a schopnost kostní dřeně na tuto stimulaci reagovat zvýšenou tvorbou granulocytů.
Leukemoidní reakce je reaktivní stav, při kterém je významně zvýšený počet leukocytů v krvi, a ty současně vykazují určité znaky předčasného vyplavení do krve, jsou „méně zralé“. U granulocytů převažuje nižší segmentace jádra, zvýší se zastoupení tzv. tyček, buněk s nesegmentovaným podlouhlým jádrem. Část bílých krvinek v cirkulaci může mít i morfologii podobnou dřeňovým metamyelocytům a myelocytům. V tom se může leukemoidní reakce podobat některým leukémiím. Na rozdíl od leukémií, které jsou nádorovým onemocněním s patologickou krvetvorbou primárně způsobenou změnou genomu (mutacemi), je leukemoidní reakce výrazem velmi intenzivní stimulace granulocytopoezy příslušnými růstovými faktory (G-CSF, GM-CSF aj.). Je to proto stav přechodný, reaktivní.
Zvýšení počtu leukocytů u leukémií je probráno v příslušné kapitole. V případě chronické myeloidní leukémie mohou být patologické (leukemické) granulocyty poměrně vyzrálé. Na rozdíl od leukemoidní reakce však mají velmi nízkou aktivitu alkalické fosfatázy a jejich funkce v imunitních obraných reakcích mohou být defektní
Jedná se o enzym hexózo-monofosfátového zkratu, který je v erytrocytu zdrojem redukovaného glutationu, antioxidačního činidla, které snižuje poškozování hemoglobinu a erytrocytové membrány kyslíkovými radikály. Gen pro G-6-PD je na X-chromosomu, proto se klinická manifestace jeho funkčně významné mutace projevuje silněji u mužů (hemizygotů).
!!!začátek petitu!!!
Protože je normální hematopoeza polyklonální a její kmenové buňky vznikají v období před náhodnou inaktivací jednoho ze dvou X-chromosomů, mají heterozygotní ženy v krvi jak erytrocyty normální, tak i erytrocyty s porušenou funkcí G-6-PD, a to v různém poměru. U žen se tato choroba většinou klinicky neprojevuje. Přesto může být u některých heterozygotních žen zkrácena délka života erytrocytů po oxidačním stresu.
!!!konec petitu!!!
Existuje velký počet různých funkčně významných mutací genu pro G-6-PD, vedle jeho dvou polymorfních forem B a A s normální funkcí. Funkčně významné mutace se častěji vyskytují u Afričanů pocházejících z centrální Afriky, u obyvatel pocházejících z některých středomořských oblastí (např. Sardinie) a u obyvatel jižní Číny28.
Klinická závažnost hemolytické anémie je velmi různorodá, v závislosti na koncentraci a vlastnostech patologické formy G-6-PD. Většinou je hemolýza vyprovokovaná zvýšeným oxidačním stresem po požití některých látek. Známé jsou hemolytické reakce na požití bobů z Vicia fava, tzv. favismus. Z léků jsou uváděna některá antimalarika, sulfonamidy, nitrofurantoin aj.
!!!začátek petitu!!!
V erytrocytech se vyskytují Heinzova tělíska, která jsou částečně z erytrocytů odstraňována s částí buněčné membrány a cytoplazmy, a to makrofágy při průchodu erytrocytů slezinou. Vznikají tak patologické formy erytrocytů označované jako „okousané buňky“.
!!!konec petitu!!!
Přestože existují případy chronické hemolytické anémie, častější jsou záchvaty hemolýzy, které mohou být velmi intenzivní, ale mají tzv. samy-sebe-limitující („autolimitující“) průběh. Hemolýze totiž přednostně podlehnou starší erytrocyty, ve kterých aktivita G-6-PD přirozeně klesá až na polovinu. Po proběhlé hemolýze zůstanou v cirkulaci mladší erytrocyty, odolnější vůči poškození oxidačním stresem. Akutní záchvat hemolýzy je spojen s hemoglobinémií, vyčerpáním haptoglobinu, hemoglobinurií a se vzestupem nekonjugovaného bilirubinu.
Soudí se, že velké rozšíření této mutace, podobně jako hemoglobinu S, nastalo v populacích vystavených vysokému riziku parazitárního onemocnění malárií. Náchylnost erytrocytů k hemolýze narušuje normální cyklus parazita ve stadiu merozoitu.
Obr. 1.15 Metabolismus glukózy v erytrocytech
Obr. 1.13 Kinetika železa v organismu. A – normální stav; B – při několikanásobně zesílené erytropoeze při chronické hemolytické anémii; C – při chronickém krvácení
Erytroblasty ztrácejí schopnost transkripce genomu vyvržením buněčného jádra ve stadiu ortochromatického erytroblastu. Vzniklé retikulocyty obsahují mitochondrie a ribosomální proteinsyntetizující aparát včetně některých mRNA. Během dozrávání retikulocytu v erytrocyt, což je proces trvající 3–4 dny, dojde ke ztrátě ribosomů a mRNA, mitochondrií27 a buňka přestane přijímat železo (protože ztratí transferinové receptory). Metabolismus zralého erytrocytu je proto oproti jiným buňkám značně zjednodušený a plní hlavně čtyři významné funkce: produkuje ATP anaerobní glykolýzou a udržuje tím v činnosti Na+/K+-ATPázu, produkuje redukovanou formu glutationu, redukuje Fe3+ methemoglobinu na Fe2+ hemoglobinu a je zdrojem 2,3-difosfoglycerátu. Všechny tyto funkce souvisejí s anaerobním metabolismem glukózy, jak je znázorněno na obrázku 1.17.
!!!obr. 1.17.!!!
Metabolické defekty, které porušují funkci erytrocytů, jsou následkem enzymových poruch glukózového metabolismu erytrocytů. Erytrocyty normálně žijí 120 dnů a během této doby nemají, na rozdíl od jiných buněk, možnost obměnit a doplnit svou enzymovou výbavu. Proto se některé enzymové defekty projeví specificky postižením erytrocytů, přestože mohou být přítomny i v jiných buňkách. Postižení erytrocytů se nejčastěji projeví jejich hemolýzou, která může být záchvatovitá, nebo chronická. Některé enzymové defekty jsou velmi vzácné, jiné jsou u příslušníků některých etnik relativně časté. Dědičnost je většinou recesivního charakteru, protože poloviční množství určitého enzymu většinou dostačuje k udržení potřebného erytrocytového metabolismu.
Obr. 1.12 Vývoj anémie při chronicky zvýšených ztrátách erytrocytů. A – normální stav; B – erytrocyty setrvávají v cirkulaci kratší dobu, proto jich zaniká víc než v normálním stavu a jejich počet v krvi se snižuje; C – anémie se stabilizuje v novém rovnovážném stavu mezi zánikem a produkcí erytrocytů (přestože erytrocyty setrvávají v cirkulaci kratší dobu, absolutní množství zanikajících erytrocytů je normální a vyrovnané s produkcí, protože jejich celkový počet v krvi je malý); D – anémie prostřednictvím erytropoetinu zvýšila produkci erytrocytů, a proto se celkový počet erytrocytů v krvi opět zvýší, přestože setrvávají v krvi kratší dobu (stav „kompenzované anémie“)
Počet všech bílých krvinek (leukocytů) v jednom litru krve je 4–10 x 109. Nižší hodnoty se označují jako leukopenie a vyšší hodnoty jako leukocytóza (viz obr. 1.6). Mezi leukocyty je nejvíce neutrofilních granulocytů – asi 70 % ze všech leukocytů. Snížení jejich počtu se označuje jako granulocytopenie (též neutropenie) nebo až agranulocytóza; zvýšení jako granulocytóza (též neutrofilie). Změny výskytu v krvi se mohou týkat lymfocytů, kterých je normálně mezi všemi leukocyty asi 20 %. Lymfocytopenie nebo lymfocytóza jsou označení používaná při příslušné změně absolutních počtů lymfocytů v krvi. Zvýšený výskyt eozinofilních granulocytů se označuje jako eozinofilie, bazofilních granulocytů jako bazofilie.
Vytvoření pevné vazby mezi granulocyty a endotelem, které předchází diapedezi granulocytů do okolní tkáně, se uskutečňuje prostřednictví membránových molekul označovaných jako CD18 a CD11a/CD11b. Ty jsou součástí β2-membránových integrinů granulocytů a monocytů. Jejich mutace jsou podkladem syndromů, při kterých je v krvi leukocytóza a současně se vyskytují časté bakteriální a mykotické infekce.
!!!začátek petitu!!!
V současné době jsou popsány 3 syndromy: 1. LAD 1 způsobený mutací genu kódující β2-integrin (CD18), 2. LAD 2 způsobený mutací genu SLC35C1, která vede k deficitu ligandů na povrchu neutrofilních granulocytů nezbytných pro vazbu selektinů na povrchu endotelových buněk, 3. LAD 3 způsobený mutacemi genů FERMT3 nebo RASGRP2, které vedou k narušení fukce β-integrinů.
!!!konec petitu!!!
Leukocytóza je způsobena zmenšením nebo chyběním marginálního poolu granulocytů. Neschopnost koncentrování granulocytů (i monocytů) v místech infekce vysvětluje častý výskyt infekcí a jejich těžký průběh. Zhoršený je také průběh hojení ran, a to i v případech, kdy nejsou infikovány. Onemocnění se dědí autozomálně recesivním způsobem.
)
Tabulka 1.12 Přehled hemolytických anémií
Korpuskulární příčina hemolýzy (většinou vrozené hemolytické anémie)
Extrakorpuskulární příčina hemolýzy:
V případě hemolytických anémií se jedná se o heterogenní skupinu onemocnění, protože příčin zvýšené hemolýzy může být mnoho. Z didaktického hlediska je vhodné dělení hemolytických anémií do dvou skupin (Tab. 1.12):
!!!tab. 1.12.!!!
Hemolytické anémie mají některé obecné patofyziologické znaky, které jsou významné pro porozumění jejich klinickým projevům.
K předčasnému zániku erytrocytů hemolýzou může převážně docházet ve slezině, event. v játrech a kostní dřeni, za účasti makrofágů, tedy extravaskulárně. Druhou možností je rozpad erytrocytů v cirkulaci, tedy intravaskulárně.
Při extravaskulární hemolýze se železo a globin reutilizují a hem je konvertován v bilirubin. Hladina nepřímého, nekonjugovaného bilirubinu proto může být zvýšena, nevzniká deficit železa a nedochází k hemoglobinurii ani hemosiderinurii. Chronicky zvýšená tvorba bilirubinu může nejen způsobovat ikterus, ale také predisponovat ke vzniku pigmentovaných žlučových konkrementů.
V případě intravaskulární hemolýzy se hemoglobin uvolněný z erytrocytů váže k haptoglobinu a tento proteinový komplex je zachycován receptory makrofágů. Tím je zabráněno ztrátám hemoglobinu a železa, ke kterým by jinak došlo následkem průniku hemoglobinu přes glomerulární membránu v ledvinách. Kapacita haptoglobinu vázat uvolněný hemoglobin je omezena jeho fyziologickou koncentrací v plazmě (asi 1 g/l plazmy) a omezenou schopností jater spotřebovaný haptoglobin doplňovat. V případně rozsáhlé intravaskulární hemolýzy může být kapacita haptoglobinu přesažena, uvolněný hemoglobin se v plazmě rozpadá na globinový dimer αβ s molekulovou hmotností asi 33 kDa, který poměrně snadno prochází glomerulární membránou. Vzniká pak hemoglobinurie. Protože je část hemoglobinu fagocytována tubulárními buňkami, ve kterých se hemoglobin změní v hemosiderin, objevuje se se zpožděním 3–4 dnů hemosiderinurie, která pak může dlouho přetrvávat. Hemolýza je v tomto případě provázena ztrátami železa. Současně může dojít i k poškození tubulárních funkcí nefronu. Koncentrace haptoglobinu se v takovém případě v plazmě významně sníží a může i chybět. Po akutně proběhlé hemolýze přetrvává nízká hladina haptoglobinu po několik dnů, než ho jaterní tkáň doplní. Stanovení koncentrace haptoglobinu je proto diagnosticky využíváno k potvrzení podezření na proběhlou intravaskulární hemolýzu. Dalším znakem svědčícím pro intravaskulárně proběhlou hemolýzu erytrocytů je zvýšení hladiny laktát-dehydrogenázy, jejího erytrocytového izoenzymu, v plazmě.
Dalším rysem hemolytických anémií je využití velké kompenzační schopnosti krvetvorné tkáně nahrazovat zanikající erytrocyty. V organismu je v těchto případech většinou dostatek železa, které se přednostně reutilizuje pro potřeby erytropoezy. Zvláště při chronických hemolytických anémiích může dojít k celkové expanzi erytroidní složky krvetvorné tkáně (erytroblasty zde normálně představují asi 20 % ze všech buněk). Krvetvorná tkáň v takových případech může dlouhodobě produkovat až osminásobek normální produkce erytrocytů. Teoreticky tedy ani zkrácení délky života erytrocytu jen na 15 dnů (z normální hodnoty 120 dnů) nemusí způsobit zjevnou anémii. Takový stav je označován jako „kompenzovaná anémie“ nebo „kompenzovaný hemolytický syndrom“. V krvi je zvýšený počet retikulocytů. Důležité je však to, že počet erytrocytů je v takovém případě velmi citlivý ke snížení produkce erytrocytů. To pak vede k rychlému rozvoji anémie.
!!!začátek petitu!!!
Přechodný pokles výkonnosti erytropoezy může způsobit jinak běžné infekční onemocnění, specificky pak infekce parvovirem B19. Rychlé zhoršení kompenzované anémie s nálezem nízkého počtu erytroblastů v kostní dřeni a retikulocytů v krvi bývá označováno jako aplastická krize. Ta může komplikovat předtím stabilizovanou, více nebo méně kompenzovanou, hemolytickou anémii.
!!!konec petitu!!!
27Erytrocyty přenášejí kyslík buňkám s oxidačním typem energetického metabolismu, ale samy kyslík nespotřebovávají.
Nejčastější je nedostatek pyruvát-kinázy. Erytrocyty trpí nedostatkem ATP a ztrátou K+. Klinický obraz hemolytické anémie je velmi variabilní.
!!!začátek petitu!!!
Bylo popsáno velké množství různých mutací pyruvát-kinázy. Dědičnost má charakter autosomálně recesivní, je proto zesilována příbuzenskými sňatky. Z důvodu velkého počtu mutací genu pro pyruvát-kinázu, jsou manifestně nemocní jedinci nejčastěji smíšenými heterozygoty se dvěma různými patologickými alelami genu pro pyruvát-kinázu. Hemolytická anémie, která se vysvětluje zvýšenou rigiditou erytrocytů a jejich sekvestrací makrofágy sleziny, bývá také označována jako nesférocytová hereditární hemolytická anémie.
!!!konec petitu!!!
!!!začátek petitu!!!
U Chédiakova-Higashiho syndromu je porušená funkce lysozomů a fagolysozomů. Porucha se proto týká nejen chemotaxe, ale i destrukce bakterií ve fagocytech. V cytoplazmě neutrofilů, monocytů a lymfocytů jsou přítomna obrovská granula. Postiženy jsou však všechny buňky, proto se vedle častých, zvláště pyogenních (hnisavých) infekcí vyskytují i jiné poruchy: albinismus, progresivní periferní neuropatie a u některých nemocných mentální retardace. Onemocnění vykazuje autosomálně recesivní typ dědičnosti.
!!!konec petitu!!!
Erytrocyt je během asi 200 000 průchodů velkým a malým krevním oběhem vystaven možnosti mechanického traumatu především v mikrocirkulaci. K tomu může dojít při deformaci průsvitu arteriol a kapilár mikrotromby (vlákny fibrinu nebo agregáty trombocytů), případně jinými vlivy. Intenzivní intravaskulární hemolýza obvykle provází trombotickou trombopenickou purpuru (TTP) a tzv. hemolyticko-uremický syndrom. V případě diseminované intravaskulární koagulace (DIC) je intravaskulární hemolýza mechanicky poškozovaných erytrocytů vyjádřena klinicky u asi čtvrtiny postižených.
!!!začátek petitu!!!
Intravaskulární hemolýza, přičítaná mechanickému poškozování erytrocytů, se může také vyskytnout při tzv. maligní hypertenzi, eklampsii a během odhojování transplantovaného orgánu. Ve všech těchto případech se předpokládá významná role endotelového poškození.
!!!konec petitu!!!
Jedná se buď o vrozené, nebo získané poruchy, které snižují schopnost granulocytů přichytit se k endotelu, migrovat ve tkáních, fagocytovat nebo destruovat fagocytovaný materiál.
!!!začátek petitu!!!
Chronická granulomatóza. Jedná se o vzácné vrozené onemocnění, při kterém nejsou neutrofily a monocyty schopné zabít fagocytované bakterie. Granulocyty a monocyty jsou defektní ve schopnosti vytvářet velké množství kyslíkových radikálů (superoxidu a peroxidu vodíku a odvozených radikálů), protože některá podjednotka membránové NADPH-oxidázy je postižena hypomorfní nebo amorfní mutací. Onemocnění vykazuje recesivní dědičnost buď autozomální, nebo vázanou na X-chromozom. Rozdílný typ dědičnosti u tohoto monogenně podmíněného dědičného onemocnění je způsoben skutečností, že mutace čtyř různých genů kódujících podjednotky výše uvedeného enzymového komplexu působí stejnou funkční poruchu.
Onemocnění se projevuje slizničními a kožními infekcemi, jakož i záněty plic, jater a jiných orgánů a tkání. Infekce jsou nejčastěji způsobeny bakteriálními patogeny, které mají ve své enzymové výbavě katalázu, a degradují proto peroxid vodíku (Staphylococcus aureus, Aspergillus sp. aj.). Tyto infekce jsou provázeny nadměrnou, ale léčebně málo účinnou, zánětlivou reakcí. Jejím projevem může být také tvorba granulomů.
Defekt myeloperoxidázy. Myeloperoxidáza je enzym granulocytů, který vytváří z peroxidu vodíku a chloridových aniontů kyselinu chlornou (HOCl). Z té vzniká chlornan OCl- a volný chlor, další látky se silnými antimikrobiálními účinky. Defekt myeloperoxidázy, který se dědí autosomálně recesivně, se nemusí projevit poruchami obrany proti infekci
, protože tento baktericidní defekt granulocytů může být kompenzován zvýšenou tvorbou kyslíkových radikálů NADPH-oxidázou.
!!!konec petitu!!!
Některé bakteriální exotoxiny obsahující fosfolipázu a některé hadí a pavoučí toxiny mohou také poškodit membránu erytrocytů. Z bakteriálních infekcí se jedná především o sepsi Clostridium welchii.
!!!začátek petitu!!!
Mírná hemolýza může provázet bakteriémii pnemokokovou, stafylokokovou, streptokokovou, E. coli a Haemophilus influenzae typu B.
Měď může vyvolat hemolýzu erytrocytů u hemodialyzovaných pacientů nebo u pacientů s Wilsonovou chorobou.
Rozsáhlé popáleniny (III. stupně a postihující 15–65 % povrchu těla) mohou být provázeny tepelným poškozením erytrocytů způsobujícím hemoglobinémii a hemoglobinurii.
K poškození erytrocytové membrány může také dojít při jaterní cirhóze provázené přítomností patologické formy lipoproteinu s nízkou denzitou (LDL), který má vysoký obsah neesterifikovaného cholesterolu. V membráně erytrocytů se zvýší obsah cholesterolu a ubyde fosfolipidů, to snižuje fluiditu jejich membrány a zvyšuje rigiditu erytrocytů. Charakteristicky se také mění jejich tvar v krevním nátěru v tzv. trnité buňky (spur cells). Ty jsou zvýšeně zadržovány a hemolyzovány ve slezině.
!!!konec petitu!!!
Hemolýzu erytrocytů způsobuje jejich napadení vývojovou formou parazitů malárie (merozoity). Merozoity od Plasmodium falciparum napadají erytrocyty bez rozdílu jejich stáří, zatímco merozoity Plasmodium vivax, ovale napadají jen retikulocyty a merozoity Plasmodium malariae pouze zralé erytrocyty. Merozoity používají hemoglobin a jiné proteiny erytrocytu pro svou výživu. Infekce erytrocytů merozoity Plasmodium falciparum mění jejich membránu, kterou se pak infikované erytrocyty přichycují k endotelu postkapilárních venul (cytoadherence) a k neinfikovaným erytrocytům (rozetování). Tím dochází k obliteraci mikrocirkulace, což může způsobit poruchy neurologické povahy nebo ischémii v myokardu. Přestože malárie může být provázena anémií, která může být chronická nebo akutní, onemocnění většinou dominuje řada jiných klinických projevů.
Hemolytické anémie, jejichž příčina leží převážně mimo vlastní erytrocyt, se nazývají extrakorpuskulárními. Jsou většinou získané.
Získané poruchy funkce granulocytů se týkají především leukémií. Funkce granulocytů však mohou být sníženy u novorozenců a u některých patologických stavů, např. u diabetes mellitus, u hemodialyzovaných pacientů, u sepse, u některých virových infekcí, u systémového lupus erythematodes, u revmatoidní artritidy, u popálenin a těžké malnutrice.
Zkrácené přežívání erytrocytů je u autoimunitní hemolytické anémie (AIHA) způsobeno přítomností autoprotilátek proti různým povrchovým erytrocytárním antigenům. Podle toho, za jakých podmínek dochází k optimální vazbě autoprotilátek na antigeny erytrocytů se AIHA klasifikuje jako: AIHA s přítomností tepelných protilátek: vážou se k erytrocytům při 37 °C a a bývají třídy IgG, AIHA s přítomností chladových protilátek: vážou se k erytrocytům při < 37 °C a většinou bývají třídy IgM, nebo smíšená AIHA s přítomností tepelných i chladových protilátek.
!!!začátek petitu!!!
Podle toho, zda je AIHA diagnostikována současně s dalším onemocněním, které způsobuje produkci antierytrocytárních protilátek lze dále AIHA s přítomností určitého typu protilátek dělit na idiopatické (bez přidruženého onemocnění) nebo sekundární (AIHA je komplikací nebo manifestací jiného základního onemocnění). Mezi taková onemocnění patří např. lymfoproliferativní onemocnění, systémový lupus erythematodes, nespecifické střevní záněty (např. ulcerózní kolitida, Crohnova choroba), nádorová onemocnění (např. tumory ovaria), některá infekční onemocnění (mycoplasma pneumoniae, EBV, syfilis, atd).
!!!konec petitu!!!
Erytrocyty s navázanými tepelnými protilátkami třídy IgG jsou nejčastěji destruovány makrofágy sleziny, které mají receptory pro Fc fragment těžkého imunoglobulinového řetězce. Makrofágy mohou erytrocyty buď celé fagocytovat, nebo jen zbavit části buněčné membrány. V tomto případě se erytrocyty mění ve sférocyty (mikrosférocyty).
K navázání chladových protilátek třídy IgM dochází typicky v chladnějších akrálních částech těla. Erytrocyty s navázanými chladovými protilátkami třídy M díky velikosti IgM (pentamer) navzájem aglutinují v cirkulaci (v postkapilárních vénách) a mohou zde způsobit částečnou nebo úplnou obliteraci mikrocirkulace. Protože k tomuto dochází při nižší teplotě, postiženy bývají akrální části těla (prsty, ušní boltce, nos). Toto se pak projeví jako akrocyanóza. Pentamer IgM váže a aktivuje C1 složku komplement (mnohem efektivněji ve srovnání s IgG). K hemolýze pak může dojít dvojím způsobem. Buď je komplement aktivován včetně vzniku terminálního lytického komplexu a dochází tak k intravaskulární hemolýze v akrálních částech těla. Nebo dojde k aktivaci komplementu pouze do složky C3b, která je navázána na erytrocytech a ty jsou pak fagocytovány makrofágy v játrech a slezině (mají receptor pro C3b složku komplementu). V tomto druhém případě dochází k extravaskulární hemolýze ve slezině a játrech.
Některé léky (penicilin, cefalosporiny, α-methyldopa amfotericin B, diclofenac) mohou způsobit AIHA tím, že navodí tvorbu autoprotilátek proti antigenům erytrocytární membrány. Pro diagnostiku antierytrocytových protilátek má základní důležitost antiglobulinový, tzv. Coombsův test. Při něm je pomocí protilátek proti lidským imunoglobulinům zjišťována jejich přítomnost na erytrocytové membráně (přímý Coombsův test), event. volných antierytrocytových protilátek v plazmě (nepřímý Coombsův test). V druhém případě jsou panelové erytrocyty nejdříve inkubovány s testovanou plazmou a pak je s nimi proveden přímý Coombsův test.
Zatímco přímý Coombsův test má především význam v diagnostice hemolytických anémií způsobených protilátkami, nepřímý Coombsův test má význam především ve zjišťování možné přítomnosti získaných protilátek proti erytrocytům u osob, kterým je opakovaně podávána transfúze krve nebo erymasy.
Tab. 1.15 Příčiny splenomegalie
Portální hypertenze
Expanze slezinných makrofágů
Expanze lymfatické složky sleziny
Extramedulární hematopoeza nebo myeloidní leukémie
* chronická myeloidní leukémie
Zvětšení sleziny se označuje jako splenomegalie. Její příčiny lze rozdělit do několika skupin (Tab. 1.15).
!!!tab. 1.15.!!!
Odstraňování většího množství erytrocytů ve slezině způsobí hyperplazii jejího makrofágového systému, což je hlavní příčinou jejího zvětšení za těchto stavů (tzv. pracovní hypertrofie).
Druhou funkční složkou sleziny je lymfatická tkáň v podobě lymfatických folikulů obsahujících převážně B-buňky. Lymfatická složka sleziny reaguje na různé antigenní podněty, a slezina se proto může zvětšit především při různých bakteriálních a houbových infekcích (např. infekční endokarditida), ale i při infekcích virových (např. infekční mononukleóza) a parazitárních, jakož i při antigenní stimulaci neinfekčního původu (např. při sérové nemoci, systémovém lupus erythematodes).
Slezina se často stává zdrojem patologické krvetvorby při akutní a chronické leukémii myeloidní nebo lymfoidní, jakož i při lymfomech a při pravé polycytémii (polycythaemia vera).
Slezina zůstává potenciálním místem krvetvorby v případech jejího postupného zániku v kostní dřeni při myelofibróze a osteopetróze. Tato extramedulární hematopoeza může také způsobit zvětšení sleziny.
Slezina je svou cirkulací součástí portálního systému. Při portální hypertenzi se velikost sleziny pravidelně zvětšuje. Splenomegalie se proto vyskytuje při jaterní cirhóze, při pravostranném srdečním selhání, při trombóze hepatálních vén (Buddův-Chiariho syndrom).
Další příčinou zvětšení sleziny jsou některé vrozené nebo získané poruchy metabolismu, při kterých se v makrofágovém systému sleziny ukládají látky, které tyto makrofágy nedokonale odbourávají. Jedná se o amyloidózu, Gaucherovu nemoc, Niemannovu-Pickovu nemoc aj.
Existují dále případy, kdy se nepodaří žádnou zřejmou příčinu zvětšení sleziny zjistit. Tyto stavy se označují jako idiopatická splenomegalie.
V tabulce 1.16 jsou uvedeny hlavní příčiny zvětšení lymfatických uzlin.
!!!tab. 1.16.!!!
O patofyziologických aspektech krevní transfúze je pojednáno v samostatné kapitole. Zde jsou uvedeny jen případné hemolytické komplikace.
V případě inkompatibility izoaglutininů a antigenů sytému AB0 může dojít k akutní hemolytické reakci. Tato reakce především hrozí, pokud krev příjemce obsahuje izoaglutininy proti erytrocytům dárce. Podkladem akutní hemolytické reakce je intravaskulární hemolýza provázená hemoglobinémií, hemoglobinurií a dalšími jejími znaky. Reakce obvykle zahrnuje další příznaky, jako je arteriální hypotenze, tachykardie, bolest na hrudníku a v bederní oblasti, zvýšení teploty, třesavka. Vedle těchto časných příznaků, které se většinou projeví již během transfúze, je nebezpečí akutního selhání ledvin a diseminované intravaskulární koagulace. Podobná akutní reakce může nastat i při převodu krve, kompatibilní v systému AB0, příjemci, který se předchozími transfúzemi senzibilizoval proti antigenům Rh, Kell nebo Duffy.
Opožděná hemolytická reakce se může dostavit za jeden až dva týdny po transfúzi, která byla provedena nemocnému, jenž se dříve senzibilizoval proti některému převáděnému antigenu (Kell, Duffy aj.), avšak titr jeho protilátek je v době transfúze nízký, takže se neprojeví při předtransfúzním vyšetření. Anamnestická imunitní reakce může vést k vytvoření dostatečného množství protilátek proti erytrocytům dárce. Erytrocyty dárce s navázanými protilátkami IgG pak mohou být z oběhu rychle odstraněny extravaskulárně, zachycením makrofágy sleziny.
Hemolýza erytrocytů Rh+ fétu, který se vyvíjí v těle matky Rh-, která již dříve donosila dítě Rh+ a vytvořila si protilátky proti tomuto erytrocytovému antigenu, je známa jako nozologická jednotka fetální erytroblastóza.
Zdrojem transplantovaných buněk je kostní dřeně, krev ev. pupečníková krev. Pokud je zdrojem buněk pacient, jde o transplantace autologní. Pokud je zdrojem příbuzný nebo nepříbuzný dárce, s dostatečnou shodou v histokopatibilních znacích, jde o transplantace alogenní nebo haploidentické. Je-li dárcem jednovaječné dvojče, jde o trensplantaci syngenní, z hlediska imunologické kompatibility blízké transplntaci autologní.
Indikacemi jsou především nádorové (akutní myeloidní leukémie, akutní lymfoblastová leukémie, myelodysplatický syndrom aj.), ale i vrozené nenádorové (např. talasémie , poruchy imunity) onemocnění krvetvorné tkáně. Transplantace kostní dřeně jsou však prováděné i za účelem léčby těžkých a progresivních autoimunitních chorob a jako součást agresivní léčby nádorů.
Transplantaci krvetvorných buněk předchází „přípravný režim“, spočívající v podání kombinace cytostatik, případně s celotělovým ozářením. Přípravné režimy podávané při autologní a alogenní transplantaci se významně liší. Stejně tak se liší přípravné režimy pro jednotlivá onemocnění, pro která je transplantace prováděna.
Přípravný režim slouží jednak k eradikaci nádorových buněk (v případě alogenní i autologní transplantace) a jednak k utlumení imunitního systému příjemce (v případě alogenní transplantace).
!!!začátek petitu!!!
Samotná transplantace spočívá v infúzí suzpenze buněk obsahujících kmenové buňky do krevního oběhu. Kmenové buňky se procesem označovaným jako uhnízdění (homing) dostávají z krve do krvetvorné tkáně, kde se ve vhodných místech uchytí a stimulovány k proliferaci, sebeobnově a diferenciaci30.
Uchycení kmenových buněk (angl. engraftment) zahrnuje i kompetici transplantovaných kmenových buněk s kmenovými bunňkami příjemce, které případně přežily po příparveném cytotoredukční předtransplantační přípravě. Výsledkem alogenní transplantace může být úplný (100%) chimerismus, kdy všechny krevní buńky jsou od dárce. Výsledkem může být i částečný chimerismus, kdy část krevních buněk pochází z transplantovaných buněk a část z přeživších kmenových buněk příjemce.
!!!konec petitu!!!
Přihojení a diferenciace uchycených kmenových buněk vyžaduje určitý čas, než z nich začnou být produkovány zralé krevní buňky. V případě, kdy byla předtransplantační přípravou zničena krvetvorná tkáň příjemce, představuje tato doba kritické období, vyznačující se leukopenií (< 0,2 x 109/l, tj. < 200/μl ) a trombocytopenií (< 0,2 x 1011/l, tj. < 20 000/μl), které jsou spojeny s rizikem infekce a krvácení. Při autologních transplantacích trvá toto období obvykle 10–15 dnů, při alogenních transplantacích 14–20 dnů. V případě leukopenie lze toto období poněkud zkrátit podáváním růstového faktoru G-CSF. Trombocytopenie většinou vyžaduje opakované transfúze koncentrátů trombocytů.
Při alogenních transplantacích může dojít k nepřijetí nebo odhojení transplantátu (rejekce) v důsledku reakce imunitního systému příjemce proti alogennímu transplantátu (H-v-G reakce; Host-versus-Graft reakce).
!!!začátek petitu!!!
Vyskytuje se častěji tehdy, nebyl-li v rámci předtransplantační přípravy dostatečně suprimován imunitní systém příjemce nebo z povahy onemocnění, pro kterou je alogenní transplantace prováděna (např. vyšší riziko u aplastické anémie). Transfúzní terapie předcházející transplantaci kostní dřeně také zvyšuje riziko neuchycení transplantátu, protože imunitní systém budoucího příjemce kostní dřeně je stimulován proti velkému spektru aloantigenů.
!!!konec petitu!!!
Reakce štěpu proti hostiteli (G-v-H reakce; Graft-versus-Host reakce) je pravidelná komplikace alogenní transplantace kostní dřeně, ke které dochází i v případě plné shody v HLA antigenech, a to z důvodů neshody v minoritních transplantačních antigenech. Rozdíl ve více než jednom HLA antigenu velmi zvyšuje riziko závažné reakce štěpu proti hostiteli. Tkáňové poškození je při této reakci způsobeno reakcí T-lymfocytů (pomocných CD4+, cytotoxických CD8+) a NK-buněk obsažených v transplantátu proti antigenům příjemce. Má akutní nebo chronický průběh. Poškozována je řada buněk a tkání: endotel, kůže, játra aj., ale také nádorové buňky, které případně zbyly po léčbě předcházející transplantaci.
29Je pozoruhodné, že se po splenektomii neprodlouží délka života normálních erytrocytů nad 120 dnů. Funkci sleziny při odstraňování starých erytrocytů a recyklaci aminokyselin a železa pak začnou plnit játra a kostní dřeň.
Transfúzí, s výjimkou autotransfúzí (autologních transfúzí – viz dále), jsou vždy do organismu vpraveny cizorodé antigeny, aloantigeny. Leukocyty a trombocyty jsou zdrojem cizorodých antigenů ze systému HLA. Erytrocyty kompatibilní s příjemcem v AB0 a Rh-systému jsou zdrojem řady dalších antigenů patřících k systému Lewis, I, P, MNSsU, Kell, Duffy, Kidd a dalším, vůči kterým se typizace rutinně neprovádí. Zdrojem cizorodých antigenů jsou i polymorfní proteiny krevní plazmy. V případě trombocytárních koncentrátů, jakož i v případě proteinových koncentrátů získaných z plazmy, jsou převáděny antigeny pocházející od více dárců, a příjemce je proto imunizován vůči velkému spektru lidských antigenů.
!!!začátek petitu!!!
Slezina představuje orgán o hmotnosti asi 150 g u mladého dospělého člověka. Ve vysokém věku se hmotnost sleziny snižuje až o jednu třetinu. Průtok krve slezinou je relativně intenzivní: 3–4 % ze srdečního výdeje, tj. asi 150 ml krve/min (300 litrů krve/den). Asi jedno promile lidí nemá slezinu a asi 20 % lidí má vedle normální sleziny ještě akcesorní slezinu.
!!!konec petitu!!!
Hlavní funkce sleziny je jednak tvorba protilátek a vyzrávání B lymfocytů a filtrace krevních elementů, zvláště erytrocytů, a jejich odstraňování z oběhu, sníží-li se jejich deformabilita nebo mají-li na svém povrchu navázané protilátky.
!!!začátek petitu!!!
Lymfatická tkáň představuje asi 15–35 % z celkové hmotnosti sleziny. Lymfatická tkáň sleziny je organizována jednak do periarteriolárních pochev a jednak do lymfatických folikulů. Z celkového množství lymfocytů přísluší asi 60 % k B-buňkám a 25 % k T-buňkám.
!!!konec petitu!!!
Hlavní funkce sleziny spočívá v odstraňování starých a poškozených erytrocytů z krve (denně asi 20 ml erytromasy)29, recyklaci aminokyselin globinu a železa hemu a v tvorbě bilirubinu. Této funkci je uzpůsobena cirkulace krve, při které se podstatná část erytrocytů nedostává do venózní části cirkulace průchodem kapilárami, nýbrž ze sinusů odtéká do venul mezibuněčnými štěrbinami s bohatým výskytem tkáňových makrofágů. Tyto makrofágy zachycují staré erytrocyty, jakož i erytrocyty patologicky změněné s nižší deformabilitou nebo nesoucí na svém povrchu protilátky IgG nebo složky komplementu C3a a C4a.
!!!začátek petitu!!!
Ne vždy přitom dochází k fagocytóze celých erytrocytů. Makrofágy mohou odstraňovat z erytrocytu jen část jeho membrány a cytoplazmy obsahující precipitovaný hemoglobin (Heinzova tělíska), zbytky nukleových kyselin (Howellova-Jollyho tělíska) nebo parazity (např. Plasmodium malariae).
!!!konec petitu!!!
Zvětšení sleziny se může projevit poruchami shrnovanými pod pojem hypersplenismus. Slezina přirozeně zadržuje až 30 % trombocytů přítomných v krvi a zadržuje též určité množství granulocytů. Tyto funkce mohou být při jejím zvětšení vystupňovány natolik, že způsobují trombocytopenii a granulocytopenii (leukopenii).
Riziko přenosu virové nebo bakteriální infekce transfúzí (např. virus hepatitidy typu C a B, HIV) je minimalizováno řadou opatření.
Transfúze 400 ml krve nebo odpovídajícího množství erytrocytů znamená příjem asi 200–250 mg železa (při jeho celkovém množství 4000–5000 mg). Pokud nedochází ke krvácení nebo k intravaskulární hemolýze, zůstává prakticky veškeré toto železo v organismu. Závažné poruchy z přetížení tkání organismu železem vznikají, zvýší-li se následkem mnoha opakovaných transfúzí významně celkové množství železa. Pravidelně se rozvinuté a ireverzibilní poškození (především jater, Langerhansových ostrůvků pankreatu a myokardu) objevuje při zvýšení celkového množství železa v organismu na 20 000 mg a více. To odpovídá asi 100 transfúzím.
Asi u 20 % lidí existuje akcesorní slezina, takže splenektomie, provedená např. z důvodu posttraumatické ruptury sleziny a nitrobřišního krvácení, nemusí vyvolat změny, které jinak mohou odstranění veškeré slezinné tkáně provázet. Mezi tyto změny patří: přechodné (někdy i trvalé) zvýšení trombocytů v krvi (trombocytémie), přechodné zvýšení granulocytů v krvi a některé erytrocyty mohou obsahovat barvitelné struktury, jako jsou Howellova-Jollyho tělíska aj. Splenektomie významně zvyšuje riziko infekčních onemocnění (zejména se jedná o infekční onemocnění způsobená opouzdřenými mikroorganismy: Streptococcus pneumoniae, Neisseria meningitidis, Haemophilus influenzae) a zhoršuje jejich průběh z důvodu určitého imunodeficitu. Z těchto důvodů je po splenektomii doporučená vakcinace proti těmto infekčním agens.
30Proces uhnízdění je představou, pro kterou existuje málo konkrétních údajů. Chemotakticky by na kmenové buňky mohl působit cytokin SDF-1 rozpoznávající receptor kmenových buněk CXCR4.
Přenosová kapacita krve pro kyslík se zvyšuje přibližně lineárně se zvyšující se koncentrací hemoglobinu v krvi. Platí to však jen v případě převodu čerstvé erymasy. V krvi skladované při teplotě 4 °C se snižuje obsah 2,3-DPG v erytrocytech a následkem toho se zvyšuje afinita krve ke kyslíku. Takové erytrocyty přenášejí kyslík méně účinně než erytrocyty normální. Trvá více než 24 hodin, než se koncentrace 2,3-DPG v transfundovaných a déle skladovaných erytrocytech upraví a zlepší se tím i jejich schopnost přenášet kyslík. Zvýšení přenosové kapacity krve pro kyslík zvýšením koncentrace hemoglobinu po transfúzi utlumí tvorbu erytropoetinu, a tedy i stimulaci krvetvorné tkáně příjemce transfúze k tvorbě vlastních erytrocytů.
Transfúze transfúzních přípravků a krevních derivátů - erymasy, koncentrátu trombocytů, koncentrátu granulocytů (indikace zcela výjimečná), plazmy, koagulačních faktorů, imunoglobulinů a jiných faktorů izolovaných z krevní plazmy, je používána v řadě indikací. Jejím primárním účelem může být doplnění krevního objemu, zlepšení dodávky kyslíku tkáním, snížení krvácení nebo zastavení krvácení, zvýšení odolnosti vůči bakteriálním a mykotickým infekcím a některé jiné indikace.
Autotransfúze vlastní (autologní) krve (většinou se podává autologní erymasa), získané před plánovanou operací (u které lze očekávat velkou ztrátu krve) opakovanými odběry a skladováním vlastní krve, je postup, který eliminuje řadu výše uvedených rizik.
Akutní poškození plic, tzv. syndrom TRALI (Transfusion Related Acute Lung Injury) patří mezi nejzávažnější potransfúzní reakce. Jedná se o formu syndromu akutního respiračního selhání (ARDS). Syndrom TRALI byl popsán v souvislosti s podáváním řady transfúzních přípravků. Nejrizikovější je plazma a transfúzní přípravky obsahující alespoň 50 ml plazmy. V patogenezi se popisuje několik mechanismů vzniku TRALI.
!!!začátek petitu!!!
Významnou roli hrají antileukocytární protilátky dárce (přítomné v transfúzním přípravku), které reagují s příslušnými antigeny na povrchu leukocytů příjemce a způsobí jejich adhezi na endotelové buňky s následnou aktivací či jejich agregaci v plicních kapilárách. TRALI vzniká rychle - do 6 hodin po podání transfúzního přípravku.
!!!konec petitu!!!
(New Engl. J. Med., 1998, 339, p. 2005–2011)
49letá žena s trombotickou trombocytopenickou purpurou (viz kazuistiku K1.4) byla léčena výměnnou transfúzí plazmy (plazmaferézou) s použitím čerstvé (zmrazené) plazmy. Asi za hodinu po zahájení plazmaferézy a za 15 minut po začátku transfúze erytromasy se snížil krevní tlak na 80/50 mmHg. Dostavila se zimnice a akutně vzniklá dušnost. Saturace krve (hemoglobinu) kyslíkem se snížila ze 100 % na 75 % během 45 minut, přestože pacientka inhalovala maskou kyslík. Parciální tlak kyslíku v arteriální krvi (paO2) byl 6,5 kPa (49 mmHg), paCO2 byl 4,6 kPa (35 mmHg) a pH krve bylo 7,48. Pacientka byla intubována a ventilace plic byla prováděna se 100% kyslíkem (FiO2 = 1). Tlak kyslíku v krvi se zvýšil na 33,0 kPa (254 mmHg) (saturace hemoglobinu kyslíkem pochopitelně dosáhla 100 %), paCO2 byl 4,8 kPa (37 mmHg) a pH krve 7,39. Rentgen plic ukázal difúzní oboustrannou konsolidaci plicní tkáně odpovídající obrazu plicního edému. Kontrola kompatibility převáděných krevních derivátů neprokázala chybu. Plazmaferéza byla dokončena s bilancí: 5990 ml dodaných tekutin a 4193 ml odebrané plazmy. Plazmaferéza pak byla prováděna i v následujících dnech, až se počet trombocytů zvýšil na 173 000/μl (1,73 x 1011/l).
Patofyziologický rozbor
Plicní parenchym je plochou asi 100 m2 v kontaktu se vzduchem a plochou asi 80 m2 v kontaktu s krví. Jemná alveolokapilární membrána může být poškozena patologickými agens, které se dostanou do plic ventilací ze vzduchu nebo krví. V tomto případě akutně vzniklého plicního selhání, které bylo z časových důvodů dáváno do souvislosti s transfúzí plazmy nebo erytromasy, musíme předpokládat, že došlo k poškození alveolo-kapilární membrány krevní cestou.
Akutní poškození plicního parenchymu krevní cestou je nejčastěji vysvětlováno aktivací granulocytů a poškozením alveolo-kapilární membrány uvolněnými proteázami, kyslíkovými radikály, tumor nekrotizujícím faktorem a dalšími látkami. V plicních kapilárách je přítomna značná část marginálního poolu leukocytů (granulocytů). K jejich aktivaci může dojít, v souvislosti s transfúzí, při přítomnosti protilátek proti leukocytům („leukoantigenům“, HLA antigenům) v plazmě obsažené v transfúzi. Významnou úlohu přitom má aktivace komplementu, kdy složka komplementu C5a zvyšuje sekvestraci neutrofilních granulocytů v plicní cirkulaci.
Vznik akutního plicního edému by mohl být při infúzní (transfúzní) léčbě způsoben také hypervolémií a selháváním levého srdce. V takovém případě by byl zvýšen centrální venózní tlak a tzv. tlak v zaklínění (viz kap. 2.2.1.4.3). U posttransfúzního akutního poškození plic jsou tyto tlaky normální.
Komentář
Parciální tlak kyslíku v arteriální krvi 33 kPa je „zbytečně vysoký“, k plné saturaci hemoglobinu kyslíkem dostačuje tlak 13 kPa. Bylo by proto možné snížit procentuální zastoupení kyslíku ve vdechovaném vzduchu, a tím snížit jeho toxické působení na plicní tkáň.
Akutní selhání plic vzniklé v souvislosti s transfúzí se vyskytuje vzácně. Ve většině popsaných případů se vyvinulo během prvních dvou hodin po zahájení transfúze. Přestože se jedná o proces odpovídající syndromu ARDS (viz kap. 3.6.1.10), je prognóza pacientů s posttransfúzním poškozením plic významně lepší než u nemocných s ARDS z jiných příčin. To ukazuje na určitou zvláštnost tohoto plicního poškození. Ve většině posttransfúzních imunitních reakcí jsou příčinou protilátky přítomné v plazmě příjemce a reagující s antigenem přítomným v použitém transfúzním přípravku. V případě akutního posttransfúzního plicního poškození je tomu naopak. Předpokládanou příčinou jsou protilátky přítomné v transfúzním přípravku. Proto byly tyto posttransfúzní komplikace popsány v případech, kdy bylo s transfúzí převedeno více než 60 ml plazmy. Pokud vznikl převáděný derivát plazmy zpracováním plazmy od velkého počtu dárců, nebyly popsány případy akutního poškození plic (titr případných protilátek je nízký, protože je „rozředěn“ plazmou od mnoha dalších dárců, kde tyto protilátky obsaženy nejsou).
Jedná se jednak o krvácivé stavy, při kterých dochází k úniku krve z cévního systému, a jednak o trombofilní stavy, které se vyznačují tvorbou krevních sraženin (trombů) v cirkulaci. Všechny probírané kategorie krvácivých stavů a trombofilních stavů mohou být vrozené, nebo získané.
Jedná se o častou komplikaci při alogenní transplantaci kostní dřeně. V případě podání transfúzního přípravku je tato komplikace vzácná. V patogenezi se uplatní T-lymfocyty dárce (obsažené v transfúzním přípravku), které reagují na HLA antigeny příjemce. Příznaky, mezi které patří kožní vyrážka, průjem, známky poškození jater aj., se objeví za 1-6 týdnů po podání transfúze. Tato komplikace, která může mít i smrtelný průběh, hrozí při transfúzi přípravku od pokrevních příbuzných a při transfúzích pacientům s imunodeficitem. Účinnou prevencí je podání přípravku zbaveného leukocytů nebo vystavení transfúzního přípravku ionizujícímu záření v dávce nad 25 Gy.
Transfúze nedostatečně temperované tekutiny, zvláště centrálním katetrem do blízkosti pravé srdeční síně, může způsobit poruchy srdeční excitability a převodu vzruchu a vyvolat srdeční arytmie.
Postransfúzní komplikace způsobené hemolýzou erytrocytů jsou probrány v pojednání o hemolytických anémiích.
Mírné zvýšení tělesné teploty, jakož i možná alergická reakce (projevující se kožní reakcí – urticaria, svědění, bolest hlavy aj.) nemusí být po transfúzi způsobeny hemolýzou. Přičítají se blíže nespecifikovaným imunitním reakcím mezi antigeny a protilátkami přítomnými v plazmě příjemce nebo dárce. K anafylaktické reakci v takovém případě může dojít po převodu několika mililitrů krve nebo plazmy.
46V případě proteinu C se jedná o komplex s trombomodulinem, trombinem a proteinem S na fosfolipidové části endotelové membrány.
Trombózou rozumíme vytváření krevních sraženin (trombů) uvnitř cirkulačního systému přichycených k cévní stěně nebo k endokardu. Hyperkoagulačním stavem rozumíme zvýšenou náchylnost ke vzniku trombů. Tromboembolií označujeme zanesení krevní sraženiny proudem krve do vzdáleného místa, kde dojde k jejímu zachycení, které omezuje průtok krve. Lokalizace trombů - arteriální, venózní, mikrocirkulace - pak určuje specifické komplikace (např. trombus lokalizovaný v hlubokém žilním systému dolních končetin může vést k plicní embolii; trombus, který vznikne v koronální arterii, může způsobit akutní infarkt myokadu, atd.)
!!!začátek petitu!!!
Koagulace krevní plazmy je dynamický děj, který je nejen iniciován a potencován, ale i inhibován. Značný potenciál inhibičních dějů demonstruje skutečnost, že naplno rozběhnutý koagulační děj v místě narušení cévního systému zůstává omezen na toto místo a negeneralizuje se.
!!!konec petitu!!!
Existuje více mechanismů, které mohou zabránit nebo utlumit srážení krve. Porucha těchto mechanismů je jedna z příčin zvýšeného rizika vzniku trombů. Přirozené antikoagulační faktory zahrnují, vedle povrchových vlastností endotelií a proudění krve, specifický endotelový protein trombomodulin a plazmatické proteiny jaterního původu: protein C, protein S a antitrombin III (viz obr. 1.26).
Trombomodulin je protein vytvářený v endoteliích a je exprimovaný na jejich buněčné membráně. Váže k sobě trombin, cílový enzym koagulační kaskády s normální substrátovou specificitou vůči fibrinogenu. Vazbou k trombomodulinu se mění substrátová specificita trombinu, který místo fibrinogenu začne preferovat protein C (plazmatický protein vytvářený játry a procházející posttranslační modifikací v podobě γ-karboxylace zbytků glutamové kyseliny, která podmiňuje jeho schopnost vytvářet komplexy s jinými faktory na fosfolipidových površích46). Enzymovým komplexem trombin-trombomodulin proteolyticky upravený protein C se sám stává aktivní proteázou, aktivovaným proteinem C, se substrátovou specificitou vůči faktorům VIII a V. To jsou významné akcelerační (potenciační) faktory prokoagulační a jejich účinnost je proteolytickým štěpením aktivovaným proteinem C snižována. Jako kofaktor působí přitom protein S. Protein S je plazmatický protein jaterního původu, jehož biologická aktivita je též podmíněna na vitaminu K závislou γ-karboxylací zbytků glutamové kyseliny (viz obr. 1.25).
Antitrombin (dříve antitrombin III) je dalším přirozeným antikoagulačním faktorem, který inhibuje účinnost prokoagulačních faktorů trombinu (IIa), faktoru Xa a faktoru IXa (a v menší míře také XIa a XIIa). Antitrombin III je protein vytvářený v játrech. Váže se k povrchu endotelií prostřednictvím zde přítomných, heparinu podobných látek.
Stavy, které zvyšují riziko vzniku trombů, lze rozdělit na vrozené a získané. Přehledně jsou uvedeny v tabulce 1.19.
!!!tab. 1.19.!!!
Nejčastější vrozenou příčinou hyperkoagulačních stavů jsou mutace faktoru V, které působí jeho rezistenci k proteolytickému štěpení aktivovaným proteinem C.
!!!začátek petitu!!!
Většinou se jedná o mutaci, při které je v molekule faktoru V zaměněná aminokyselina arginin (v poloze 506) za glutamin. Tato mutace se označuje jako leidenská (Leiden, město v Nizozemí). Odhaduje se, že jejími nositeli (heterozygoty) jsou asi 3 % populace. Heterozygotní stav zvyšuje riziko tromboembolické nemoci 7krát, homozygotní stav 20krát. Současný výskyt jiného vrozeného nebo získaného defektu, který zvyšuje krevní srážlivost, dále zvyšuje toto riziko.
!!!konec petitu!!!
V důsledku působení zevních nebo vnitřních mechanických sil na stěnu cév (poranění, krevní tlak, působení gravitačních sil, vliv svalové práce) může dojít k jejímu narušení, které umožní únik části krve do extravaskulárního prostoru. Rozsah krvácení může být od mikroskopického krvácení po masivní krvácení, akutně ohrožujících život. Rozlišuje se krvácení vnitřní a zevní. Nebezpečí krvácení může být nejen v množství uniklé krve, ale v některých případech také v jeho lokalizaci. Vnitřní krvácení může narušit důležité strukturní vztahy (např. arteriální krvácení v mozku) a vyvolat následné efekty (tlakové změny ve tkáni mohou způsobit ischémii, zvýšení tvorby kyslíkových radikálů v místě krvácení při pozdější degradaci hemu, kdy se uvolňuje Fe2+) nebo může narušit významnou funkci (např. průběh srdeční diastoly při krvácení do perikardiální dutiny). V této kapitole se budeme zabývat stavy, které zvyšují riziko úniku krve z cévního systému. Hemoragické diatézy jsou způsobené buď zvýšenou fragilitou cévní stěny, nebo sníženou schopností krve zacelovat místa narušení cévního systému. V obou případech těchto poruch může být jejich příčinou buď vrozený, nebo získaný patologický stav.
!!!začátek petitu!!!
Zástavy krvácení a udržování tekutosti krve se účastní cévní endotel, subendotelové struktury, hladké svalstvo cévní stěny, trombocyty, koagulační faktory (aktivátory a inhibitory) a krevní proud, který je aktivním účastníkem těchto dějů.
Endotelie zamezují srážení krve několika mechanismy. Ve své membráně (podobně jako normální krevní buňky) neobsahují přístupný tkáňový faktor. Na zevní straně buněčné membrány mají negativní elektrický náboj. Exprimují trombomodulin a heparinu podobné látky. Zvyšují cévní průsvit produkcí NO a prostaglandinu I2 (PGI2), které mají inhibující účinek na trombocyty. Jsou zdrojem tkáňového aktivátoru plazminogenu (tPA) (viz obr. 1.27). Zároveň jsou dynamickou strukturou, která jim, zvláště po jejich aktivaci, umožňuje interakci s trombocyty prostřednictvím adhezivní molekuly PECAM (CD31). Po aktivaci mohou též vázat a zadržovat leukocyty a umožnit jejich diapedezi do extravaskulárního prostoru. Po vystavení endotoxinu (lipopolysacharidu z Gram-negativních bakterií) a cytokinu TNF mohou také exprimovat ve své membráně tkáňový faktor.
Neaktivované trombocyty, které jsou normálně přítomny v krvi v množství od 150 000 do 400 000/μl (1,5–4 x 1011/l), mají receptory pro kolagen, fibronektin, von Willebrandův faktor, vitronektin, laminin, trombin, ADP, tromboxan A2, PECAM (CD31) a některé jiné molekuly.
Aktivaci trombocytů vyvolá jejich vazba na kolagen, fibronektin, vitronektin, laminin a von Willebrandův faktor, působení trombinu (koagulačního faktoru IIa) za účasti Ca2+, adrenalinu a ADP. Ve svých granulech obsahují řadu účinných látek, které mohou po své aktivaci rychle uvolnit do okolí (obr. 1.18). Aktivace trombocytů proto vyvolá řadu místních reakcí včetně aktivace dalších trombocytů (pozitivní zpětná vazba). Aktivované trombocyty začnou syntetizovat další účinné látky (prostaglandiny, tromboxan A2 aj.). Aktivované trombocyty také zvýšeně exprimují glykoproteiny IIb-IIIa, které představují receptor pro fibrinogen. Prostřednictvím tohoto receptoru, plazmatického fibrinogenu a destičkového fibrinogenu uvolňovaného po jejich aktivaci se trombocyty váží navzájem, což se označuje jako agregace trombocytů (Obr. 1.19).
!!!obr. 1.18. a 1.19.!!!
Koagulační faktory (aktivátory a inhibitory) jsou přítomné v krevní plazmě v neaktivní formě tzv. zymogenů (proenzymů). Jedná se o potenciální proteázy, které ze zymogenů vznikají jejich specifickým proteolytickým štěpením. Toto štěpení provádějí aktivované formy jiných koagulačních faktorů, čímž vzniká aktivační kaskáda. Někdy dochází k samoštěpení autokatalýzou (např. faktor IIa štěpí a aktivuje faktor II – protrombin). Cílovým proteolytickým enzymem v této kaskádě je trombin – faktor IIa. Vedle aktivace a autoaktivace se v řízení koagulačních dějů uplatňuje i jejich inhibice substrátem, který nemůže být rozštěpen, změna jejich substrátové specificity a proteolytické štěpení prokoagulačních faktorů. Jedná se tedy o komplexní děj, jehož průběh může být ovlivněn řadou faktorů. Koagulace normálně vyústí v tvorbu fibrinové sítě a její následnou fibrinolýzu. Odhaduje se, že jeden mililitr plazmy obsahuje koagulační potenciál dostatečný pro konverzi veškerého fibrinogenu na fibrin, tj. pro koagulaci 5 litrů krve.
Průtok krve je významný tím, že účinně snižuje koncentraci prokoagulačních faktorů v určitém místě cirkulačního aparátu. Zpomalení průtoku krve nebo změna proudění krve z laminární formy v turbulentní obecně zvyšuje tendenci ke vzniku krevní sraženiny. Naproti tomu silný průtok krve místem poranění cévní stěny může účinně bránit vzniku krevní sraženiny.
!!!konec petitu!!!
Fyziologický průběh zástavy krvácení zahrnuje dva následné děje, které se označují jako primární hemostáza a sekundární hemostáza.
Obr. 1.19 Aktivace, metamorfóza a sekrece trombocytů vyvolaná kolagenem, trombinem, adrenalinem nebo ADP (hydrolýza membránových fosfolipidů, inhibice adenylátcyklázy, mobilizace intracelulárního Ca2+, fosforylace intracelulárních proteinů). Číslice označují jednotlivé enzymy: 1 – fosfolipáza A2 (fosfatidylcholin), 2 – lipáza (diacylglycerol), 3 – fosfolipáza C (fosfatidylinozitol-4,5-difosfát), 4 – cyklooxygenáza; AA – arachidonová kyselina, TxA2 – tromboxan A2, vWF – von Willebrandův faktor
Vrozené poruchy cévní stěny. Mezi vrozené poruchy cévní stěny patří teleangiectasia hereditaria haemorrhagica – morbus Rendu-Osler-Weber. Jedná se o autozomálně dominantně děděné onemocnění. Cévní stěna je v některých místech zeslabena a drobná céva je zde rozšířena. Tyto cévní defekty lze vidět prostým okem, pokud se vyskytují na kůži nebo na sliznici, což je jejich častá lokalizace. Mohou se však vyskytovat i v různých orgánech, např. v plicích. V plicích mohou být zdrojem pravo-levých plicních zkratů a způsobovat tak snížení tenze kyslíku v arteriální krvi a hypoxémii. U části nemocných dochází k drobnému krvácení do gastrointestinálního traktu a event. k hematurii.
Ehlersův-Danlosův syndrom a Marfanův syndrom jsou dva vrozené defekty pojiva. K častějšímu krvácení do kůže při drobných traumatech přispívá, vedle poruchy cévní stěny, zvýšená pohyblivost cév v dermis.
Získané poruchy cévní stěny souvisejí s poruchami pojivové tkáně.
!!!začátek petitu!!!
Tzv. senilní purpura je způsobena jednak zvýšenou cévní fragilitou ve vyšším věku a jednak zvýšeným poškozováním drobných cév následkem atrofie podkožní tkáně (úbytek elastických vláken). Petechie a purpura, které provázejí některé infekce (spála, spalničky aj.), jsou vysvětlovány poškozením cévní stěny bakteriálními exotoxiny. V případě tzv. horečky Skalistých hor parazitují rickettsie v endoteliích, které jsou tím poškozeny.
!!!konec petitu!!!
Zvýšená fragilita cév může být způsobena nedostatkem vitaminu C (ten je potřebný pro syntézu hydroxyprolinu, který je nezbytnou složkou kolagenu). Henochova-Schönleinova purpura je způsobena vaskulitidou, která zvyšuje permeabilitu cévní stěny. Poškození cévní stěny je pravděpodobně způsobené imunitími komplexy antigenu a protilátky obsahujícími IgA a aktivací komplementu. Vyskytuje se u dětí a mladých lidí, často po infekčním onemocnění respiračního aparátu.
Bodová mutace v genu kódující protrombin (G20210A) je druhým nejčastějším vrozeným trombofilním stavem. Onemocnění je autozomálně dominantně dědičné a zvyšuje riziko tromboembolické nemoci asi 2-3krát.
Vrozený nedostatek nebo snížená funkce antitrombinu (dříve antitrombin III)
Zvýšené riziko vzniku trombů je již při snížení hladiny antitrombinu pod 50 %, tedy i u heterozygotů s jednou inaktivační mutací.
!!!začátek petitu!!!
Bylo popsáno více než 130 mutací v genu kódující antitrombin, které způsobují jeho kvantitativní (typ I) nebo kvalitativní (typ II) deficit. Typ II lze dále dělit podle toho, jaká doména antitrombinu je postižena přítomnou mutací. U homozygotů je třeba předpokládat neúčinnost heparinu pro snížení krevní srážlivosti, protože účinek heparinu je podmíněn reakcí s antitrombinem (viz obr. 1.26).
!!!konec petitu!!!
Mezi získané stavy, které zvyšují riziko vzniku trombů, patří především poškození endotelové výstelky cévně-srdečního systému. Dochází k němu nejčastěji aterosklerotickými změnami, dále poraněním a zánětem (endocarditis, phlebitis). Dalším získaným faktorem, který zvyšuje riziko vzniku trombů, je zpomalení průtoku krve při srdečním selhání, hyperviskózním syndromu, imobilizaci či místních překážkách odtoku krve z tkáně nebo vznik turbulentního proudění, kdy část krve déle setrvává na určitém místě (aktivované koagulační faktory jsou z oběhu odstraňovány především při průchodu krve játry).
Dalším získaným faktorem je trombocytémie (esenciální trombocytémie nebo stavy po splenektomii) a patologická přítomnost tkáňového faktoru v cirkulaci (nádorová onemocnění, endotoxémie, sepse, poranění, porod, velké operace). Poškození jaterní činnosti způsobuje nejen nedostatek prokoagulačních faktorů, ale i nedostatek antikoagulačních faktorů a snížené odstraňování aktivovaných prokoagulačních faktorů.
Užívání perorálních antikoncepčních látek obsahujících estrogeny snižuje hladinu antitrombinu.
Klinickým projevem jsou opakované žilní trombózy a tromboembolie do plic. Mohou se vyskytnout již u mladých lidí.
Mezi vrozené, často zděděné defekty (s autozomálně dominantním typem dědičnosti), které zvyšují riziko vzniku trombů, patří nedostatek rezistence faktoru V vůči aktivovanému proteinu C, mutace protrombinu, deficit antitrombinu III, proteinu C nebo S, a některé dysfibrinogenémie.
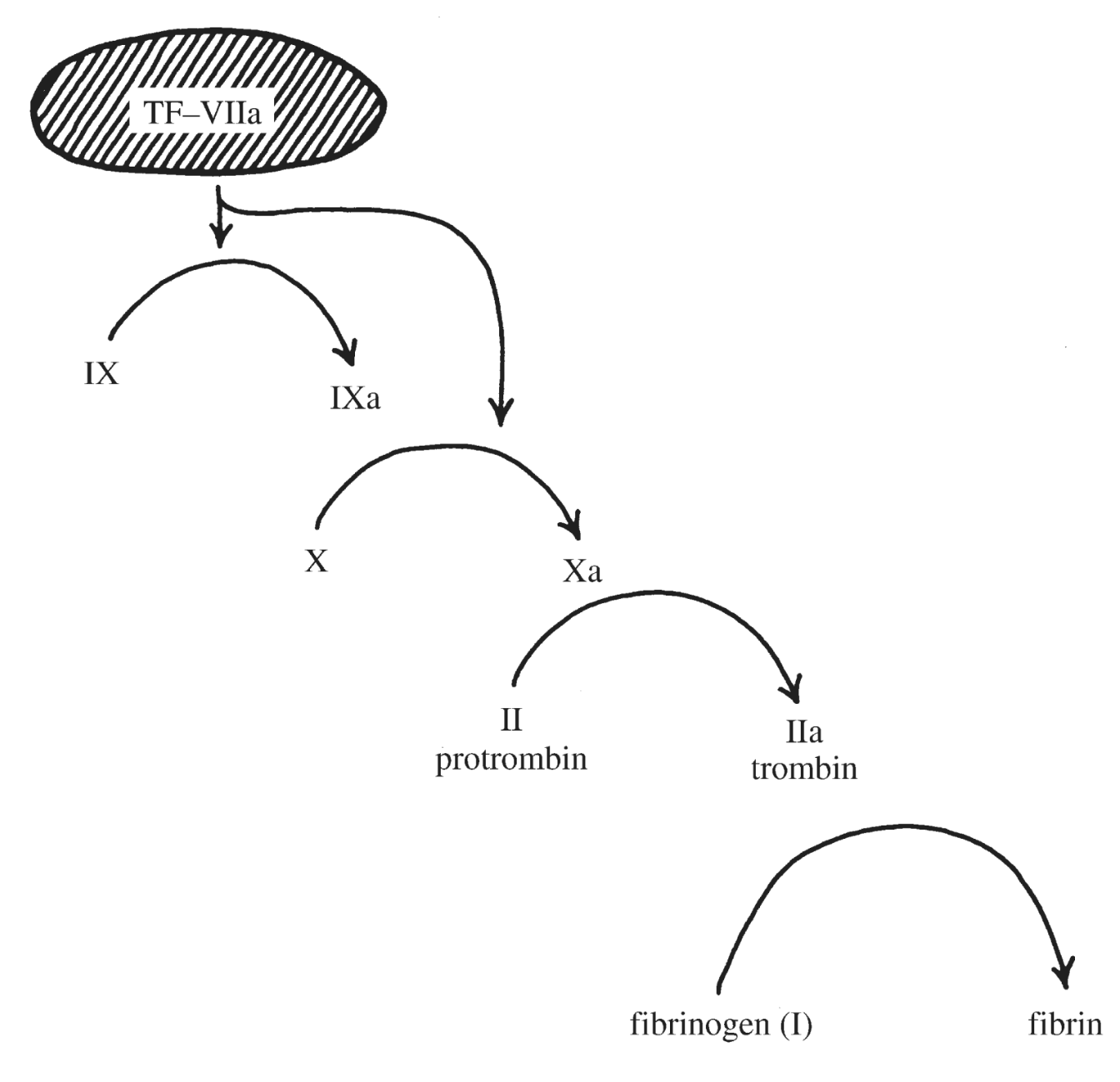
Zástavu krvácení bezprostředně zahajují reakce označované jako primární hemostáza. Jedná se o lokální vazokonstrikci zprostředkovanou jednak nervově, místním axonálním reflexem, a následně vazokonstrikčními faktory uvolněnými z aktivovaných trombocytů, zvláště tromboxanem A2 a serotoninem. Další součástí primární hemostázy je vytvoření agregátu z trombocytů spojených navzájem vazbou jejich receptorů gpIIb-IIIa k fibrinogenu uvolněnému z aktivovaných trombocytů nebo pocházejícímu z plazmy (Obr. 1.19).
Primární hemostázy se neúčastní plazmatické koagulační faktory. Významným činitelem jsou však trombocyty. Proto se s poruchami primární hemostázy setkáváme především při trombocytopenii, nebo při špatné funkci trombocytů, při trombocytopatiích.
Klinickým projevem poruch primární hemostázy je tvorba petechií.
!!!začátek petitu!!!
Petechie jsou místa v kůži nebo na sliznici, kde dojde k malému úniku krve do okolí drobné cévy, což se projeví červenou skvrnkou o velikosti menší než 2 mm. Okolí může být zažloutlé z důvodu degradace hemoglobinu. V případě náhle vzniklé těžké trombocytopenie se mohou četné petechie31 objevit především na dolních částech nohou, okolo kotníků, protože zde je cévní stěna vystavena, ve vzpřímené poloze, vedle krevního tlaku ještě hydrostatickému tlaku krevního sloupce.
!!!konec petitu!!!
K dalším klinickým příznakům svědčícím pro poruchu primární krvetvorby patří purpura32, krvácení z nosu (epistaxe)33, krvácení z dásní, krvácení do gastrointestinálního traktu, hematurie, intenzivní menstruační krvácení (menoragie).
Z výskytu těchto příznaků vyplývá, že trombocytopenie a trombocytopatie nejsou jedinou příčinou drobného krvácení do tkání, sliznic a kůže, ale že významný je i stav cévní stěny.
!!!obr. 1.19!!!
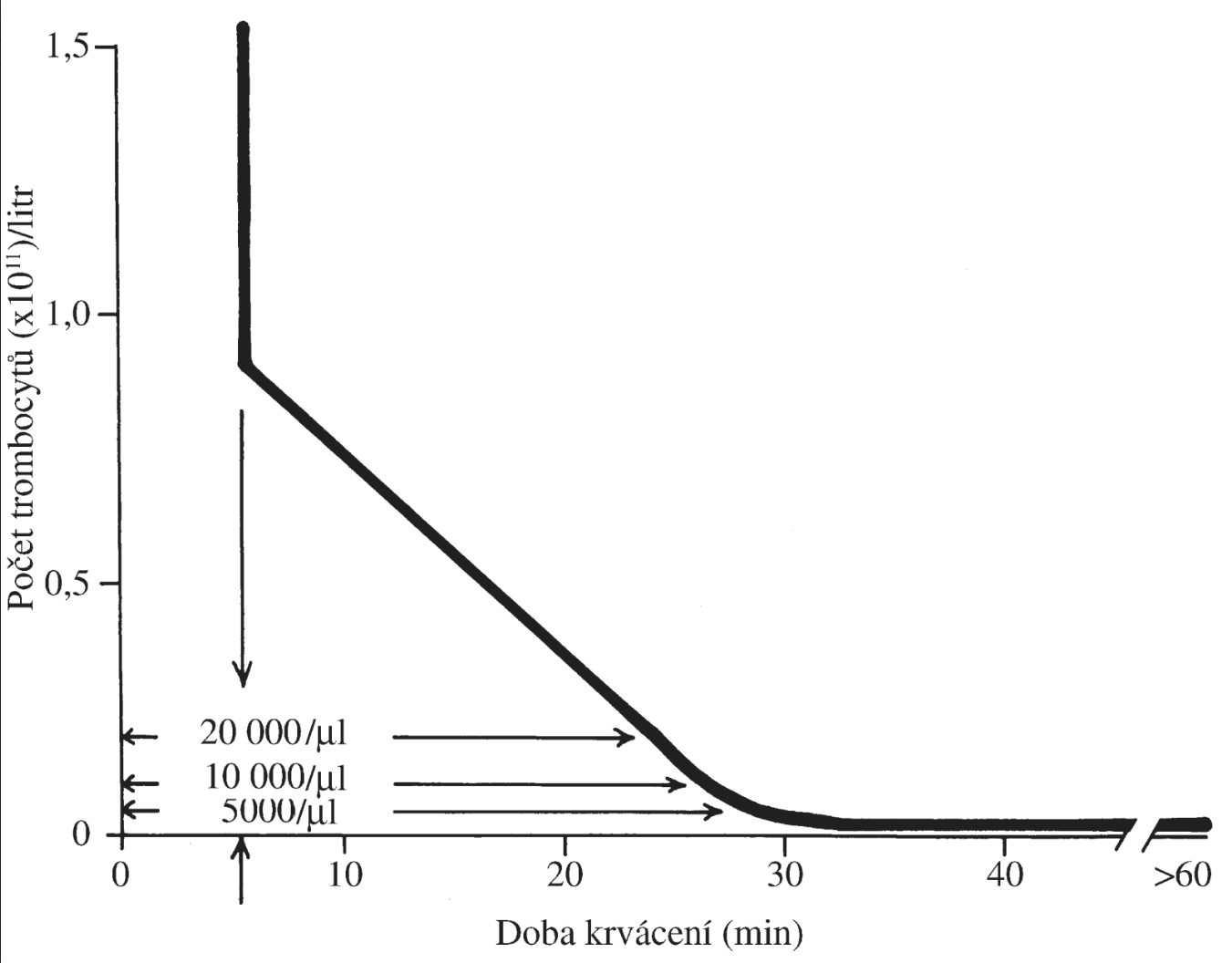
Tato je součástí aplastických a hypoplastických syndromů krvetvorné tkáně, myelodysplastického syndromu, akutní myeloidní leukémie, akutní lymfoblastická leukémie, chronické lymfocytární leukémie, některých nehodgkinských lymfomů, u kterých dochází k infiltraci kostní dřeně a útlaku normální krvetvorby. Trombocyty setrvávají v cirkulaci asi 8–10 dnů. Proto při náhle vzniklé poruše tvorby trombocytů se vyvine klinicky závažná trombocytopenie za 4–5 dnů.
Tab. 1.17 Přehled příčin trombocytopenie
Snížená produkce trombocytů
Zvýšená destrukce nebo konzumpce trombocytů
Sekvestrace trombocytů ve slezině
Vzácné dědičné onemocnění (s autosomálně recesivním typem dědičnosti) známé jako trombocytopenie s chyběním loketních kostí os radius se vyznačuje nízkým výskytem nebo chyběním megakaryocytů v kostní dřeni. Příčina může být v poruše signalizace trombopoetinového receptoru. Existují další vzácné vrozené trombocytopenie.
Mutace, které inaktivují příslušné geny, jsou účinné již při postižení jedné alely, proto má dědičnost těchto poruch charakter autozomálně dominantní. Fenotyp nedostatku proteinu C a proteinu S je stejný: opakující se venózní trombózy a tromboembolie do plic.
!!:začátek petitu!!!
Dysfibrinogenémie
Mutace v řetězcích tvořících molekulu fibrinogenu mohou snižovat jeho konverzi na fibrin působením faktoru IIa (trombinu) a způsobovat tak poruchu srážení krve. Byly však popsány i případy vyššího výskytu trombóz u pacientů s dysfibrinogenémie (zřejmě v důsledku zvýšení hladin jiných koagulačních faktorů).
!!!konec petitu!!!
38Takové složení trombu odpovídá tzv. červenému trombu, který se většinou vytváří ve venózní části cirkulace v prostředí pomalého průtoku krve. Krevní sraženiny vznikající v arteriální části cirkulace a v srdci jsou často převážně tvořeny adherujícícmi trombocyty s malým obsahem fibrinu a erytrocytů. Jsou označovány jako tzv. bílé tromby a mají jiné vlastnosti především v tom, že jsou celkově pevnější a méně podléhají fibrinolýze.
Tab. 1.18 Pozitivita tří koagulačních laboratorních testů u poruch sekundární hemostázy (koagulopatií)
| Laboratorní test | Poznámka |
Prodloužený test aPTT a test PT normální
| mírné krvácivé projevyzávažné krvácivé projevy |
Prodloužený test PT a normální test aPTT
| |
Prodloužený test aPTT i PT
| |
Prodloužený trombinový čas (TT)
| mírné krvácivé projevyzávažné krvácivé projevy |
aPTT – aktivovaný parciální protrombinový čas, PT – protrombinový čas (Quickův
39Krvácení do kloubní dutiny se označuje jako hemartróza. Ta se může
Podstatou sekundární hemostázy je koagulace krevní plazmy končící vytvořením sítě fibrinu, do které jsou zavzaty trombocyty a erytrocyty. K fibrinu adheruje plazmatický protein plazminogen38.
!!!začátek petitu!!!
Koagulopatie tedy postihují proces srážení krve ve vlastním slova smyslu. Jedná se o enzymově urychlovaný děj, probíhající optimálně při 37 °C a spočívající v kaskádě a smyčkách (pozitivní zpětné vazby) proteolytických reakcí.
!!!konec petitu!!!
Příznaky vrozených nebo získaných koagulopatií jsou shodné s příznaky poruch primární hemostázy ve výskytu častější epistaxe, krvácení do gastrointestinálního traktu, hematurie a menoragie. Liší se však od nich chyběním petechií a purpury. U koagulopatií dochází k rozsáhlým krvácením do tkání, dochází ke vzniku velkých hematomů, a to nejen v kůži a sliznicích (petechie však nejsou), ale nejednou v hlubokých tkáních, kloubech, svalech, retroperitoneu a případně i v mozku. Tvorba hematomů je způsobena narušením cévního řečiště a déletrvajícím a opakovaným krvácením do okolní tkáně.
!!!začátek petitu!!!
Pokud je zdrojem krvácení rozpoznatelné mechanické trauma, mohou hematomy vzniknout se zpožděním několika hodin, ale i dvou, tří i více dnů po poranění. Vysvětlením je malá pevnost uzávěru cévního narušení v důsledku koagulační poruchy. Ke krvácení také může dojít bez zjevného traumatu, následkem fyziologických mechanických sil působících na cévní systém, např. v oblasti kloubů nejvíce vystavených působení tělesné hmotnosti (koleno, kotník, kyčelní kloub)39. Pokud dochází v důsledku poranění k zevnímu krvácení nebo ke krvácení do hlubokých tkání a orgánů, pak komprese rány není účinným prostředkem k zástavě krvácení, které většinou pokračuje formou dlouhodobého prosakování krve. Rána může krvácet s proměnlivou intenzitou i po několik dnů. Účinným prostředkem k zástavě krvácení je dodání nedostatkového nebo nefunkčního koagulačního faktoru nebo alespoň transfúze čerstvé plazmy od zdravého dárce. Při poruchách sekundární hemostázy lze také očekávat zhoršené hojení ran, protože tento proces je zahájen vytvořením krevního koagula, uvolněním látek z trombocytů a účastí fibrinových vláken v migraci buněk.
!!!konec petitu!!!

(New Engl. J. Med., 1998, 339, p. 2005–2011)
49letá žena měla dva týdny před přijetím do nemocnice menoragii a ortostatické potíže. Den před přijetím byla provedena kyretáž dělohy a žena dostala transfúzi erytromasy. V den přijetí se dostavila náhlá intenzivní bolest hlavy a následně neschopnost mluvit, přičemž pacientka rozuměla jednoduchým pokynům. Měla pokleslý pravý ústní koutek.
Výsledky fyzikálního a laboratorního vyšetření
Při příjmu bylo zjištěno: tělesná teplota 36,8 °C, srdeční frekvence 81/min, frekvence dýchání 22/min, TK 150/80 mmHg. Pacientka byla bledá. Na hrudníku bylo několik petechií a mnohočetné pavoučkovité hemoragie. Na očním pozadí byly oboustranně patrné hemoragie a edém papily. Byla patrná chabá obrna na pravé straně obličeje a jazyk se uhýbal doprava. Další nálezy zahrnovaly dysmetrii při testu „prst na nos“, symetrické zesílení šlachových reflexů, oboustranně pozitivní příznak Babinského, mírné známky meningeálního dráždění.
Laboratorní vyšetření plazmy a moče: většina nálezů byla normálních. Byla přítomna mírná proteinurie a mírně zvýšená glykémie. Celkové plazmatické proteiny byly sníženy, především na úkor albuminu.
Krevní obraz: druhý den byla pacientka léčena transfúzí erytromasy (tab. K1.3).
Laboratorní vyšetření mozkomíšního moku získaného lumbální punkcí – viz tab. K1.4.
Patofyziologický rozbor
Anamnéza svědčí pro hemoragickou diatézu, která se projevovala menoragií (zesíleným menstruačním krvácením) a mozkovou příhodou. CT vyšetření hlavy naznačilo možnost jak ischemických, tak hemoragických ložisek v mozku. Vyšetření mozkomíšního moku svědčí pro krvácení do meningeálního prostoru.
Výskyt petechií a pavoučkovitých hemoragií na kůži svědčí pro poruchu primární hemostázy. Tomu odpovídá laboratorní nález trombocytopenie. Základní koagulační testy, Quickův test (PT) a aPTT, byly normální. Koagulační systém plazmy byl proto intaktní.
Druhým výrazným laboratorním nálezem byla přítomnost patologických forem erytrocytů, hodnocených jako schistocyty, mikrocyty a ovalocyty. Celkově byl tento nález hodnocen jako výrazná poikilocytóza. Patologické formy erytrocytů vznikají mechanickým poškozením erytrocytů při jejich průchodu mikrocirkulací obsahující mikrotromby. Anémie a zvýšená hodnota výskytu retikulocytů, spolu s patologickými formami erytrocytů, nepřímo ukazují na zvýšenou hemolýzu erytrocytů. Tato relativně akutní hemolýza je nedostatečně kompenzována zvýšenou produkcí erytrocytů (retikulocytů) krvetvornou tkání. Další vyšetření na potvrzení zvýšené hemolýzy, jako např. stanovení hladiny haptoglobinu a bilirubinu v plazmě, případně průkaz hemosiderinu v moči, nebyla pravděpodobně provedena.
Výsledky provedených vyšetření ukazují na mikroangiopatii způsobenou destičkovými a fibrinovými mikrotromby a poškozující mechanicky erytrocyty při jejich průchodu drobnými cévami. Trombocytopenie je chápána jako důsledek nadměrné spotřeby trombocytů při tvorbě mikrotrombů (proto označení „trombotická trombocytopenie“), která je nad možností krvetvorné tkáně kompenzovat tyto ztráty zvýšenou tvorbou krevních destiček. Na rozdíl od diseminované intravaskulární koagulace nedochází k významné konzumpci plazmatických koagulačních faktorů včetně fibrinogenu. Koagulační testy krevní plazmy jsou proto normální.
Komentář
Diagnóza TTP byla založena na přítomnosti trombocytopenie, poškození erytrocytů způsobujícím hemolytickou anémii s nálezem schistocytů (tzv. mikroangiopatická hemolytická anémie) a na neurologických příznacích. Další dva obvyklé příznaky, porušení funkce ledvin a zvýšená tělesná teplota, vyjádřeny nebyly.
Obr. 1.24 Srážení plazmy pokračuje i po inaktivaci komplexu tkáňového faktoru a aktivovaného faktoru VII (TF–VIIa) inhibitorem cesty tkáňového faktoru (TFPI). Srážení je udržováno proteolytickou aktivitou aktivovaného faktoru II (protrombinu)
31Toto ukazuje, že k drobným narušením integrity cévní stěny přirozeně dochází, avšak že jsou tato narušení okamžitě uzavřena destičkovými agregáty.
32Jedná se o četné drobné petechie, které splývají. Únik krve do okolí drobných cév způsobuje červené zabarvení kůže, které nezmizí účinkem komprese (stejně jako v případě petechií). V případě purpury jsou pozorovatelné kožní změny v průměru větší než 3 mm.
33Časté epistaxe mohou být také způsobeny cévní anomálií nebo překrvením nosní sliznice změněné zánětem.
Trombocytopenie mohou být vrozené (vzácně), nebo získané. Trombocytopenie je důsledkem buď nedostatečné tvorby trombocytů v krvetvorné tkáni, nebo jejich zvýšené spotřeby (při trombotické trombocytopenické purpuře), jejich sekvestrace ve slezině (při hypersplenismu), anebo jejich destrukce imunitními mechanismy (autoimunitní onemocnění) (Tab. 1.17).
!!!tab. 1.17.!!!
Ke zvýšené spotřebě trombocytů dochází při trombotické trombocytopenické purpuře (TTP). Toto závažné onemocnění patří do skupiny trombotických mikroangiopatií a vyznačuje se tvorbou trombocytárních mikrotrombů (agregátů), které vznikají současně na mnoha místech krevního oběhu (postižena je mikrocirkulace). Mikrotromby způsobují ischémii a následně poškození životně důležitých orgánů - zejména ledvin a CNS. Nedochází přitom k aktivaci plazmatického koagulačního systému. Tento proces je natolik intenzivní, že dochází ke spotřebování trombocytů s následnou těžkou trombocytopenií a rizikem krvácivých komplikací. Přítomnost mikrotrombů v cirkulaci vede k mechanickému poškozování erytrocytů a vzniku mikroangiopatické hemolytické anémie, která se vyznačuje přítomností schistocytů.
TTP vzniká v důsledku poškození endotelu a uvolnění von Willebrandova faktoru (vWf), jakož i jeho snížené proteolytická degradace buď z důvodu autoprotilátek proti proteáze ADAMTS13 štěpící von Willebrandův faktor nebo z důvodu vrozeného defektu této proteázy (Upshawův–Schulmanův syndrom)34.
!!!začátek petitu!!!
Při deficitu ADAMTS13 nedochází ke štěpení multimerů vWf s následnou pokračující adhezí trombocytů na multimery vWf prostřednictvím trombocytárních glykoproteinů GPIb-IX a tvorbě trombů v mikrocirkulaci.
!!!konec petitu!!!
Klinicky se TTP projevuje horečkou, únavou, krvácivými projevy, CNS symptomatologií (pestrá symptomatologie, může být přítomna i těžká porucha vědomí) a renálním selháním. Laboratorně je přítomna většinou těžká trombocytopenie, anémie se známkami hemolýzy a přítomností schistocytů, mohou být přítomny laboratorní známky poškození ledvin.
Na obrázku 1.20 je ukázána porucha zástavy krvácení z drobného kožního poranění ve vztahu k počtu trombocytů v krvi. Normálně dochází k zástavě krvácení průměrně za 4 minuty (a do 10 minut). Snížení počtu trombocytů pod 50 000/μl již signifikantně porušuje primární hemostázu. Významné riziko nadměrného krvácení vzniká při snížení počtu trombocytů pod 20 000/μl a mimořádně vysoké je toto riziko při snížení počtu trombocytů pod 5000/μl.
!!!obr. 1.20.!!!
!!!začátek petitu!!!
Trombocytopenie je provázena vzestupem hladiny trombopoetinu v plazmě, protože tento je konstitutivně vytvářen zejména v játrech a jeho hladina závisí na intenzitě jeho spotřeby. Trombocyty obsahují receptory (c-Mpl) pro trombopoetin a spolu s megakaryoblasty a megakaryocyty váží trombopoetin na specifické receptory a spotřebovávají ho. Jeho hladina je proto většinou v nepřímém vztahu k počtu trombocytů v cirkulaci (obr. 1.21).
Trombopoetin je glykoprotein, produkt specifického genu. Jeho receptor, produktu buněčného protoonkogenu c-mpl, je přítomen na megakaryocytech i trombocytech. Na rozdíl od erytropoetinu a erytropoetinového receptoru, které jsou nezbytné pro tvorbu erytrocytů, není trombopoetin a jeho působení pro tvorbu trombocytů zcela esenciální.
!!!konec petitu!!!
!!!obr. 1.21.!!!
Nebyly popsány patologické stavy způsobené nedostatečnou tvorbou trombopoetinu. Zdrojem trombopoetinu je více tkání, hlavně jaterní tkáň, ledviny, slezina a kostní dřeň. Tvorba trombopoetinu je popisována jako konstitutivní, tj. stálá a nepodléhající žádným známým vlivům (srovnej např. s erytropoetinem, jehož produkce naopak kriticky závisí na dostupnosti kyslíku). Přestože je v organismu vytvářeno stálé množství trombopoetinu, jeho plazmatická hladina se za některých stavů výrazně mění. Příčinou těchto změn jsou rozdíly v odstraňování trombopoetinu z cirkulace, při kterém je nejhlavnějším faktorem jeho spotřeba v trombocytech a megakaryocytech kostní dřeně. Jak megakaryocyty, tak i trombocyty mají ve své buněčné membráně specifické receptory pro trombopoetin a lze říci, že trombopoetin je spotřebováván úměrně celkovému počtu těchto receptorů. Při trombocytopenii se proto hladina trombopoetinu v krvi zvyšuje. Naopak zvýšení počtu trombocytů v krvi, např. po infúzi jejich koncentrátu nebo po splenektomii, sníží hladinu trombopoetinu (viz obr. 1.21). Působení trombopoetinu se projevuje zvýšeným počtem megakaryocytů v kostní dřeni, vyšším stupněm jejich polyploidity a zvýšením počtu trombocytů v krvi.
Zvýšeným rizikem trombózy a tromboembolické nemoci se také mohou projevovat defekty ve fibrinolýze. Je sporné, zda primární defekty ve fibrinolýze vedou ke zvýšenému výskytu arteriální nebo žilní trombózy.
!!!začátek petitu!!!
Fibrinolýza je děj, během kterého je trombus převeden do tekutého stavu. Fibrinolýza je výsledkem degradace fibrinu proteolytickým enzymem plazminem. Plazmin vzniká v krevní sraženině z plazmatické bílkoviny plazminogenu, která adheruje k vláknům fibrinu (viz obr. 1.27). Plazminogen je přeměněn v účinný plazmin proteolytickou aktivací tkáňovým plazminogenovým aktivátorem (tPA) a plazminogenovým aktivátorem urokinázového typu (uPA), jejichž zdrojem jsou endotelové buňky. Plazminu může vedle fibrinu sloužit za substrát i fibrinogen. V plazmě je však přítomen účinný inhibitor plazminu, α2-inhibitor plazminu, který zabraňuje degradaci fibrinogenu plazminem uvolněným do krve.Tvorba a uvolňování tPA endotelovými buňkami je tlumeno inhibitorem, označovaným jako inhibitor plazminogenového aktivátoru (Plasminogen Activator Inhibitor – PAI). Ten je vytvářen účinkem trombinu (faktoru IIa) na endotelové buňky. PAI zřejmě na počátku koagulačního procesu zpomaluje degradaci nově vznikajícího fibrinu. Je zřejmé, že tvorba fibrinu a jeho degradace, procesy, o kterých se soudí, že probíhají paralelně, musí mít zpočátku rovnováhu posunutou ve prospěch tvorby fibrinu a až po určité době se tato rovnováha musí změnit ve prospěch fibrinolýzy47.
!!!konec petitu!!!
40Koagulační faktory (proenzymy, zymogeny) jsou tradičně označovány římskými číslicemi. Po jejich proteolytické aktivaci jsou označovány stejnou číslicí s přidáním písmene „a“.
Koagulace krve in vivo. Proniknutí krve (plazmy) do subendotelových a extravaskulárních prostor, způsobí navázání plazmatického koagulačního faktoru VII na tkáňový faktor, protein přítomný v buněčné membráně a na povrchu většiny buněk. Po navázání na tkáňový faktor se faktor VII stane velmi citlivým k proteolytické aktivaci stopovým množstvím aktivovaných koagulačních faktorů Xa40 a VIIa, které jsou normálně přítomné v plazmě. Faktor VIIa dále aktivuje faktory IX a X. Nejedná se tedy jen o kaskádu, ale i o amplifikační smyčku (pozitivní zpětnou vazbu). Koagulace se proto rozběhne rychle a intenzivně. Výsledkem je aktivace protrombinu (faktoru II) na trombin (faktor IIa). Trombin, proteolytický enzym, je konečným enzymem v těchto reakcích (Obr. 1.22). Trombin pak ještě dále zesiluje svoji tvorbu tím, že proteolyticky aktivuje akcelerační faktory V (proakcelerin) a VIII (antihemofilický globulin), které se mění na faktory Va a VIIIa. Aktivuje i faktor IX. Mnohonásobně se tím zesílí vznik faktoru Xa. Koagulace zahájená aktivací VII. faktoru tkáňovým faktorem (dříve nazývaným tromboplastin) se stane závislou ve svém pokračování na faktorech VIIIa a IXa, protože faktor VIIa začne být inhibován tzv. inhibitorem cesty tkáňového faktoru (tissue factor pathway inhibitor – TFPI) (obr. 1.23).
!!!obr. 1.22. a 1.23.!!!
Koagulace krve in vitro. Kontakt krve s aktivačním povrchem zahajuje srážení krve aktivací XII. (Hagemannova) faktoru za účasti vysokomolekulárního kininogenu. Při této reakci se současně aktivuje prekalikrein na kalikrein (Obr. 1.24). Faktor XIIa pak aktivuje faktor XI a ten pak další faktory (Obr. 1.24), až dojde ke vzniku faktoru Xa. To je tzv. vnitřní cesta srážení krve. Ta se uplatní po styku krve (plazmy) s aktivačním povrchem (vysokomolekulární kininogen, prekalikrein a faktor XII adherují k aktivačnímu povrchu, a tím jsou přivedeny do vzájemné reakční blízkosti41,42).
!!!začátek petitu!!!
Při zástavě krvácení, po narušení celistvosti cévního systému, se in vivo aktivace faktoru XII v koagulaci krve uplatňuje velmi málo. Výzanmná je však pro tvorbu kalikreinu a dalších kininů (např. bradykininu), v aktivaci komplementu a v aktivaci plazminogenu na plazmin (který působí fibrinolýzu).
!!!konec petitu!!!
!!!obr. 1.24.!!!
V tabulce 1.18 je uvedena souvislost nedostatku určitých koagulačních faktorů s pozitivitou tří laboratorních koagulačních testů.
!!!tab. 1.18.!!!
Zdrojem plazmatických koagulačních faktorů je jaterní proteosyntéza. V játrech jsou také modifikovány zbytky glutamové kyseliny obsažené ve faktorech II (protrombin),VII, IX a X (tzv. protrombinový komplex) na di-γ-karboxyglutamovou kyselinu. Tato posttranslační změna podmiňuje jejich schopnost vazby k fosfolipidovým povrchům43 a schopnost vazby Ca2+. Její realizace je závislá na vitaminu K.4400
Také při diseminované intravaskulární koagulaci (DIC) bývá trombocytopenie. Její hlavní příčinou je konzumpce trombocytů při diseminovaném koagulačním procesu. Uvažuje se však i o možnosti mechanického poškozování trombocytů při DIC, analogického poškozování erytrocytů
Léčebné zákroky cílené na časné zprůchodnění arterií obliterovaných nově vzniklým trombem nebo tromboembolizací naruší tuto rovnováhu ve prospěch fibrinolýzy. Používá se např. proteolytického enzymu bakteriálního původu, streptokinázy. Rychlost, s jakou lze dosáhnout „rozpuštění“ trombu při těchto zákrocích, opět demonstruje ohromný potenciál prokoagulačních a antikoagulačních pochodů. Tento potenciál je většinou využit jen v malém zlomku, díky úměrně účinným kontrolním mechanismům.
Postiženo bývá více koagulačních faktorů současně, pokud není příčinou nedostatku určitého faktoru tvorba specifických neutralizačních protilátek. Mezi nejčastější příčiny patří porušení funkce jater, jaterní insuficience nebo selhání. Druhou častou příčinou je nedostatek vitaminu K, způsobený nejčastěji poruchou vstřebávání tuků v gastrointestinálním traktu. Další významná příčina je iatrogenní, terapeutické podávání antikoagulancií za účelem řízeného snížení krevní srážlivosti (warfarin, heparinové preparáty, nová perorální antikoagulancia - inhibitory trombinu, inhibitory faktoru Xa). Další poměrně častou a klinicky velmi závažnou získanou poruchou koagulace je stav označovaný jako diseminovaná intravaskulární koagulace (viz dále).
42Při koagulačním vyšetření aktivovaného parciálního tromboplastinového času (aPTT) se k plazmě přidává kaolin, který zvětšuje reakční plochu smáčivého povrchu.
34Nejúčinnějším léčebným zákrokem je výměnná transfúze plazmy (plazmaferéza).
41Při koagulačním vyšetření „srážení celé krve“ se krev odebere do zkumavky se smáčivým povrchem a udržuje se v teplotě 37°C. Měří se doba, za kterou se krev změní v koagulum (norm. 4–10 minut).
Slezina normálně obsahuje (zadržuje) asi 30 % veškerých trombocytů přítomných v krvi35. Zvětšená slezina může zadržovat natolik velké množství trombocytů, že vzniká klinicky významná trombocytopenie. Ta proto patří k základním symptomům hypersplenismu.
44Recesivní fenotyp onemocnění je podmíněn tím, že koagulační porucha se projevuje jen při velkém snížení plazmatické koncentrace daného koagulačního faktoru.
Často se jedná o recesivně děděná44 onemocnění, vázaná na X-chromozom. Příčinou však mohou být i nově vzniklé mutace příslušných genů. Odhaduje se, že nové mutace jsou odpovědné asi za jednu třetinu onemocnění. Rodinná anamnéza pak nemusí být pozitivní. Postižen bývá jen jeden koagulační faktor. Závažnost postižení, stupeň vyjádření patologického fenotypu, se však může u jednotlivých postižených značně lišit.
Nejčastějšími vrozenými poruchami koagulace vznikají v důsledku mutací genů, které kódují koagulační faktory VIII a IX, von Willebrandův faktor a faktor V.
43Vazba jiných faktorů k fosfolipidovým povrchům, vysokomolekulárního kininogenu, faktorů Va a VIIIa, se uskutečňuje jinak, na základě hydrofobních interakcí.
Trombocytopenie se může vyskytnout po implantaci umělé srdeční chlopně, při vaskulitidách a již zmíněné DIC. Soudí se, že trombocytopenie je ve všech těchto případech způsobena předčasným zánikem trombocytů v důsledku jejich poškození a spotřebování v koagulačních pochodech.
35Proto se po splenektomii obvykle přechodně zvyšuje počet trombocytů v krvi, což je provázeno mírným zvýšením rizika vzniku trombů.
Koagulační faktor VIII (antihemofilický globulin – AHG) je normálně přítomen v plazmě v množství asi 0,1 mg/l. Jedná se o poměrně velký protein s molekulovou hmotností asi 300 kD, který je značně nestabilní, pokud není (elektrostatickými silami) navázán na von Willebrandův faktor přítomný v plazmě. Důležitým, ale ne výlučným zdrojem faktoru VIII jsou játra. Je např. v granulech obsažených v trombocytech, ze kterých je secernován po jejich aktivaci. Funkce koagulačního faktoru VIII spočívá, po jeho proteolytické aktivaci na faktor VIIIa, v lokálním zvýšení koncentrace koagulačních faktorů potřebných pro vznik aktivní formy koagulačního faktoru X (faktoru Xa), aktivátoru protrombinu (Obr. 1.25). Je považován především za důležitý kofaktor účinku koagulačního faktoru IXa, který zesiluje vznik faktoru Xa, který je součástí "protrombinázy", která aktivuje velká množství protrombinu na trombin. Faktor VIII je vytvářen hepatocyty a v plazmě se vyskytuje v komplexu s von Willebrandových faktorem.
!!!obr. 1.25.!!!
Snížení množství faktoru VIII v plazmě do asi 40 % normální koncentrace nepůsobí koagulační poruchu. Proto ženy s jednou funkční alelou genu pro faktor VIII na dlouhém raménku chromosomu X a sníženou koncentrací faktoru VIII v plazmě jsou přenašečky tohoto onemocnění, avšak jen zcela výjimečně mohou být postiženy krvácivostí odpovídající hemofilii A. Snížení množství faktoru VIII na 40–5 % působí jen velmi mírné projevy onemocnění (zvýšené krvácení v souvislosti s chirurgickým výkonem nebo poraněním, nebývá spontánní krvácení), snížení na 5–1 % působí mírné onemocnění (zvýšené krvácení v souvislosti s chirurgickým výkonem nebo poraněním, nebývá spontánní krvácení) a pouze snížení hladiny tohoto faktoru pod 1 % působí závažnou koagulační poruchu.
Příčinou nízké hladiny faktoru VIII mohou mutace genu kódujícího faktor VIII nebo homozygotní forma von Willebrandovy nemoci, kdy v plazmě chybí von Willebrandův faktor, který poskytuje faktoru VIII stabilitu.
!!!začátek petitu!!!
Gen pro koagulační faktor VIII je lokalizován na dlouhém raménku chromozomu X, má 26 exonů a může být postižen řadou různých mutací (popsáno bylo >2100 mutací asociovaných s hemofilií A), včetně malých či velkých delecí, missence mutací (způsobí záměnu aminokyseliny), nonsense mutací (mění kodón pro určitou aminokyselinu na stop kodon), frameshift mutace (způsobí posun čtecího rámce), atd. U asi 45 % nemocných s hemofilií A je přítomna inverze části jeho genu lokalizovaná ve 22. intronu, která působí vznik zkrácené molekuly faktoru VIII. Značná variabilita mutací tohoto genu ztěžuje možnost jejich detekce na úrovni zjištění specifické DNA nukleotidové sekvence. Široké spektrum mutací genu faktoru VIII vysvětluje rozdíly v jejich dopadu na výslednou koncentraci faktoru VIII v plazmě a jeho funkční vlastnosti. To se pak odráží ve velmi rozdílné klinické závažnosti hemofilie A. Rozdíly v závažnosti koagulační poruchy však byly popsány i u jedinců, kteří měli identickou mutaci genu pro koagulační faktor VIII. Vysvětlení může spočívat v současném výskytu mutace jiného koagulačního faktoru, např. v časté mutaci faktoru V (leidenská mutace)45. Hemofilie A je cca v 75 % případů dědičným onemocněním, zbylých 25% případů vzniká sporadicky.
Základem terapie hemofilie A je náhrada chybějícího faktoru VIII buď v profylaktickém režimu (snižuje riziko krvácení) nebo v podání koncentrátu faktoru VIII při krvácení.
Příčinou nedostatku faktoru VIII může být také vytvoření specifických protilátek (neutralizační protilátky IgG). K tomu může dojít u hemofiliků, kterým je opakovaně podáván koncentrát tohoto faktoru (rekombinantní faktor VIII nebo faktor VIII izolovaný z plazmy dárců).
!!!konec petitu!!!
Von Willebrandova nemoc. V plazmě je paralelně snížená hladina von Willebrandova faktoru a koagulačního faktoru VIII. Aktivita koagulačního faktoru VIII v pásmu 50–20 % patří k základním znakům Willebrandovy nemoci I. typu. Jiný typ mutace genu pro von Willebrandův faktor může zrušit jeho schopnost elektrostaticky vázat faktor VIII. Ten v tomto případě může být více snížen (obvykle mezi 25 a 10 % jeho normální aktivity), ale hladina von Willebrandova faktoru je normální (typ von Willebrandova onemocnění 2N), jakož i jeho schopnost vázat aktivované trombocyty.
Obr. 1.25 Aktivace faktoru XII vyvolá tvorbu kininů a aktivuje fibrinolýzu. Aktivuje také „vnitřní systém srážení krve (plazmy)“. LMWK – kininogen o nízké molekulové hmotnosti; HMWK – kininogen o vysoké molekulové hmotnosti
Trombocyty mají dva funkčně významné glykoproteinové komplexy, gpIb-IX a gpIIb-IIIa. Těmi se mohou vázat jednak k subendotelovým strukturám (kolagenu I a II) prostřednictvím von Willebrandova faktoru a dále k plazmatickému a trombocyty secernovanému fibrinogenu. Tyto glykoproteiny plní funkci receptorů, kterými jsou realizovány procesy adheze a agregace trombocytů (viz obr. 1.19).
!!!začátek petitu!!!
Bernardův-Soulierův syndrom je způsoben vrozeným defektem komplexu gpIb-IX. Glanzmannova trombastenie je způsobena defektem komplexu gpIIb-IIIa. Obě poruchy vykazují autozomálně recesivní typ dědičnosti a obě způsobují klinicky závažnou poruchu primární hemostázy.
!!!konec petitu!!!
Von Willebrandova nemoc je spolu s hemofilií A a B nejčastějším vrozeným defektem hemostázy. Narušuje adhezi trombocytů. Nejedná se však o trombocytový defekt, protože patogeneticky působí snížená hladina von Willebrandova faktoru (vWF) v plazmě. Následkem toho je u postižených jedinců sekundárně snížená funkce trombocytů.
Von Willebrandův faktor je však rovněž plazmatickým nosičem koagulačního faktoru VIII (antihemofilický globulin). Bez vazby k von Willebrandovu faktoru je koagulační faktor VIII nestabilní, a jeho hladina v plazmě je snížená. Proto se toto onemocnění projevuje příznaky odpovídajícími poruchám jak primární hemostázy, tak i sekundární hemostázy (koagulace).
Von Willebrandův faktor je vytvářen endoteliemi a megakaryocyty. Má několik molekulárních forem a normálně je přítomen v krevní plazmě, v granulech trombocytů a subendoteliálně. Již mírné snížení koncentrace vWF v plazmě (norm. asi 10 mg/l) nebo chybění jeho vysokomolekulárních forem (multimerů) působí poruchu v adhezi trombocytů. Von Willebrandova nemoc má několik forem, většinou s autozomálně dominantním typem dědičnosti a zachovanou (ale sníženou) přítomností vWF (heterozygoti). Mezi klinické projevy patří časté krvácení z nosu a dásní, krvácení do gastrointestinálního a urogenitálního traktu, zesílené krvácení po úrazech a po operacích. Krvácení do kloubů a svalů je vzácné. Z laboratorních testů je prodloužená doba krvácení, je snížena koncentrace vWF v plazmě a je snížena aktivita faktoru VIII. Quickův test (PT) je normální.
Existuje i získaná forma von Willebrandovy nemoci, podmíněná vytvořením protilátek proti von Willebrandovu faktoru.
Imunní trombocytopenie (ITP, dříve idiopatická trombocytopenická purpura) je autoimunitní onemocnění charakterizované trombocytopenií a rizikem krvácení. Na trombocytopenii se u ITP podílí několik mechanismů.
!!!začátek petitu!!!
U asi 60 % pacientů jsou detekovatelné autoprotilátky (IgG) proti destičkovým glykoproteinům IIb-IIIa nebo Ib-IX. Trombocyty s navázanými protilátkami jsou odstraňovány z cirkulace fagocyty retikuloendotelového systému (zejména ve slezině). Dalším mechanismem je snížená produkce trombocytů - autoprotilátky se vážou na glykoproteinové receptory na povrchu megakaryocytů a negativně ovlivňují produkci trombocytů. Roli hraje také abnormální imunita zprostředkovaná T lymfocyty (deficit regulačních T lymfocytů, Treg a deregulace cytotoxických T lymfocytů,).
!!!konec petitu!!!
ITP dělíme na primární (bez zjevné příčiny) a sekundární (trombocytopenie je zprostředkována imunitně při jiném základním onemocnění). Sekundární ITP může doprovázet lymfoproliferativní onemocnění (např. chronickou lymfocytární leukémii), systémový lupus erytematodes a další autoimunitní onemocnění.
!!!začátek petitu!!!
Z hlediska trvání choroby pak rozlišujeme ITP nově diagnostikovanou (do 3 měsíců od vzniku), perzistující (v trvání od 3 do 12 měsíců) a chronickou (trvá > 12 měsíců).
!!!konec petitu!!!
Mezi klinické projevy ITP patří kožně-slizniční krvácivé projevy – petechie, ekchymózy, epistaxe, menorhagie, slizniční krvácení v dutině ústní nebo v gastrointestinálním traktu, klinicky velmi závažné krvácení do CNS. ITP se může projevit po proběhlé virové infekci. Existuje také ITP navozená léky (popsáno > 300 léků s potenciálem navodit ITP, např. nesteroidní protizánětlivé léky, trimethoprim-sulfamethoxazol a jiné). Laboratorně je u ITP patrná izolovaná trombocytopenie (často velmi těžká) bez dalších abnormalit v krevním obraze. Definitivní diagnóza ITP však spočívá zejména ve vyloučení ostatních možných příčin trombocytopenie.
Heparinem indukovaná trombocytopenie s trombózou (HITT)
vzniká u malé části pacientů, kteří dostávají nefrakcionovaný heparin (vzácněji po podání nízkomolekulárního heparinu). HITT je způsobená protilátkou proti destičkovému faktoru 4 (PF4) v komplexu s heparinem. Komplex PF4-heparin-protilátka se váže na trombocyty a působí jejich aktivaci, zvýšenou adhezi k endotelu a agregaci. Výsledkem je trombocytopenie a hyperkoagulační stav se zvýšeným rizikem vzniku trombózy.
!!!začátek petitu!!!
U HITT dochází k významnému poklesu počtu trombocytů (i když velmi těžká trombocytopeni s hodnotami <10x109/l nebývá typická) 5-10 dnů po podání heparinu. Trombóza (arteiální či venózní) se vyvine u 50% pacientů s HIT. Léčba HITT spočívá v okamžitém vysazení heparinu a zahájení antikoagulace jiným preparátem (warfarin je kontraindikován).
!!!konec petitu!!!
Jaterní tkáň je zdrojem šesti koagulačních faktorů tzv. protrombinového komplexu (vyznačujících se γ-karboxylací některých zbytků kyseliny glutamové závislou na epoxidu vitaminu K): faktorů II, VII, IX, X, proteinu C a proteinu S. Dalšími koagulačními faktory vytvářenými v játrech jsou faktor I (fibrinogen), V, XI, XII a XIII. Játra jsou také místem skladování a metabolické aktivace vitaminu K.
!!!začátek petitu!!!
Pokud je jaterní onemocnění provázeno splenomegalií a hypersplenismem, může nepřímo způsobovat trombocytopenii. U jaterních onemocnění býbá aktivována fibrinolýza, která může zhoršovat krvácivý stav. Příčinou je snížená inaktivace plazminu v játrech.
!!!konec petitu!!!
Ztráta jaterního parenchymu je proto často provázena prodloužením laboratorních testů srážení krve a případně i klinicky významnou tendencí ke krvácení. Mohou se na ní přitom podílet jak poruchy sekundární hemostázy, tak i porucha primární hemostázy.
Játra jsou však také místem, kde jsou z plazmy odstraňovány aktivované formy koagulačních faktorů, jakož i antikoagulačních faktorů: antitrombinu III, proteinu C a proteinu S (Obr. 1.26). Poškození jater a ztráta části jaterního parenchymu proto mohou zvyšovat riziko vzniku diseminované intravaskulární koagulace (viz dále) nebo systémové fibrinolýzy.
!!!obr. 1.26.!!!
Vrozený defekt trombocytů snižuje jejich aktivaci, agregaci nebo sekreci (degranulaci).
Fibrinogen (koagulační faktor I) je v plazmě přítomen v množství asi 2,5 g/l. Jedná se o dimer ze dvou komplexů sestávajících z řetězců α, β a γ (celková molekulová hmotnost 340 kDa). Fibrinogen je substrátem pro trombin, který z něj odštěpuje fibrinopeptidy A a B za vzniku fibrin-monomerů. Ty následně polymerizují ve fibrin.
Afibrinogenémie je stav, kdy v plazmě není přítomen žádný fibrinogen. Krvácivé projevy bývají závažné. V případě drobných narušení cévní stěny však bývá aktivita trombocytů dostačující k zabránění krvácivých projevů.
Dysfibrinogenémie je výsledkem různých mutací genů pro fibrinové řetězce, mutací, které mění vlastnosti fibrinogenu a způsobují sníženou srážlivost krve. Protože se většinou jedná o heterozygoty (autosomální), nebývají žádné klinické projevy, nebo jsou jen velmi mírné. Na druhé straně, některé mutace mohou způsobit zvýšenou tendenci ke konverzi fibrinogenu na fibrin. Ty proto znamenají zvýšené riziko vzniku trombóz.
Patofyziologický stav DIC ukázal na provázanost koagulačních a fibrinolytických pochodů. Jiným stavem narušení fyziologické rovnováhy koagulačních a fibrinolytických pochodů je primárně zvýšená fibrinolýza, způsobená zvýšenou aktivitou fibrinolytických pochodů.
Zvýšení fibrinolýzy mohou způsobit některé živočišné toxiny. Fibrinolýzu zvyšuje nadbytek tkáňového plazminogenového aktivátoru (tPA) např. u pacientů s cirhózou jater (tPA je normálně odstraňován z cirkulace jaterní tkání, a proto se jeho aktivita v plazmě může při úbytku jaterní tkáně zvýšit). Při primární fibrinolýze je krvácivý stav způsoben snížením hladiny fibrinogenu v krvi.
!!!začátek petitu!!!
Jinou příčinou primární fibrinolýzy může být vrozený nedostatek inhibitoru plazminového aktivátoru (PAI) nebo α2-plazminového inhibitoru.
!!!konec petitu!!!
45Leidenská mutace faktoru V může zvýšit produkci trombinu u nemocného hemofilií, a tím poruchu koagulace částečně korigovat.
Jako trombocytopatie se označují stavy, při kterých je funkce trombocytů defektní z důvodu jejich vnitřní poruchy. Tyto stavy mohou být příčinou poruch primární hemostázy i při dostatečném počtu trombocytů v krvi.
V cytopalazmě trombocytů jsou přítomna granula, v nichž je skladována řada látek (např. ADP nebo adhezivní glykoproteiny, jako fibronektin, trombospondin, von Willebrandův faktor), které jsou po jejich aktivaci uvolněny (viz obr. 1.18). Skladovací a degranulační funkce těchto granul může být vrozeně defektní. Heřmanského-Pudlákův syndrom (porucha primární hemostázy, okulokutánní albinismus a pigmentové granulace v makrofázích kostní dřeně) a Chédiakův-Higashiho syndrom představují vrozené poruchy funkce trombocytů, spočívající v defektu jejich granul a nitrobuněčné cytokinetiky a následně v jejich porušené skladovací a degranulační funkci.
Obr. 1.18 Erytrocyty ve slezině normální (A) a u nemocného s hereditární sférocytózou (B)
Gen pro koagulační faktor IX je také na dlouhém raménku chromosomu X v blízkosti genu pro faktor VIII. Mutace jsou a asi jedna třetina onemocnění je způsobena novými mutacemi (ty nemusí být dědičné). Hemofilie B je asi třikrát méně častá než hemofilie A, ale vykazuje stejný charakter dědičnosti. Faktor IX je vytvářen převážně v játrech a pro svoji aktivitu, schopnost vazby k fosfolipidovým povrchům, musí být posttranslačně karboxylován. V plazmě je přítomen v asi 100krát vyšší koncentraci než faktor VIII, tj. v koncentraci asi 10 mg/l. Podobně jako u hemofilie A také závažnost koagulační poruchy v případě hemofilie B odráží stupeň snížení hladiny faktoru IX, klinické projevy tedy rovněž vznikají jen při velkém snížení koncentrace faktoru IX a jsou pak stejné jako u hemofilie A. Základem terapie je podávání chybějícího faktoru IX.
Vitamin K je resorbován v tenkém střevě společně s tuky. Vedle potravy jsou jeho zdrojem i bakterie v tenkém střevě. V těle je skladován především v játrech. Jeho nedostatek se může projevit pozitivitou koagulačních vyšetření a poruchou sekundární hemostázy během 7–10 dnů po akutním přerušení jeho příjmu nebo vstřebávání. V jaterních mikrosomech je vitamin K epoxidován do účinné formy kofaktoru enzymové γ-karboxylace zbytků glutamové kyseliny.
Při nedostatku vitaminu K jsou v játrech vytvářeny neúčinné formy proteinů protrombinového komplexu, které nemají schopnost adherence k fosfolipidovým povrchům, což je předpoklad jejich normální funkce při srážení krve. Parenterální podání vitaminu K může příznivě ovlivnit krevní srážlivost již za 5–10 hodin.
!!!začátek petitu!!!
Dikumarolové preparáty (warfarin) a cefalosporinová antibiotika inhibují redukci a recyklaci vitaminu K a způsobují příznaky jeho nedostatku.
!!!konec petitu!!!
Obr. 1.26 Úloha fosfolipidové membrány a iontů Ca2+ v procesu srážení krve. Zvýšení koncentrace koagulačních faktorů, které umožní jejich vzájemné interakce, se uskutečňuje na fosfolipidových membránách v přítomnosti iontů Ca2+. Koagulační faktory TF (tkáňový faktor), VIIIa, Va a HMWK (vysokomolekulární kininogen) se váží k fosfolipidové membráně (např. aktivovaných trombocytů) hydrofobními vazbami. Koagulační faktory VII, IX, X a II se váží k fosfolipidové membráně glutamovou kyselinou se dvěma karboxylovými skupinami. Tato posttranslační modifikace glutamové kyseliny je závislá na vitaminu K. Při nedostatku vitaminu K mají tyto čtyři koagulační faktory sníženou funkci, nebo jsou zcela nefunkční
!!!začátek petitu!!!
Byly popsány ojedinělé rodiny s těmito defekty (převážně autozomálně recesivní typ dědičnosti). Klinické projevy byly spojeny jen s velkým krvácením (operace, velká porabnění). Defekty faktoru XII (a vysokomolekulárního kininogenu a prekalikreinu) se neprojevují krvácivým stavem, pouze laboratorní vyšetření aktivovaného parciálního tromboplastinového času (aPTT) je významně pozitivní (prodlouženo až nad 100 s).
Faktor XIII, fibrin-stabilizující faktor, je enzym (transglutamináza), který zpevňuje fibrinovou strukturu vytvářením příčných vazeb a činí ji tím odolnější vůči fibrinolýze. Vzniklá sraženina je nestabilní a je urychleně rozpuštěna fibrinolytickým procesem s následným obnovením krvácení po 24–48 hodinách po poranění.
!!!konec petitu!!!
Při diseminované intravaskulární koagulaci (DIC) se primárně jedná o spuštěnou koagulaci, která následně, vyčerpáním koagulačních faktorů, způsobuje zvýšenou krvácivost. Proto jsou tyto stavy výstižně charakterizovány jako konzumpční koagulopatie. Koagulace krve je normálně výhradně lokální fenomén, omezený na místo úniku krve z cévního řečiště. V případě DIC je koagulační proces iniciován a nedostatečně kontrolován na mnoha místech v cévním řečišti, aniž by toto bylo v počátečních fázích nezbytně narušeno.
Uvnitř cévního systému není normálně přítomen tkáňový faktor. Začátek DIC spočívá v aktivaci faktoru VII tkáňovým faktorem, který je patologicky přítomen uvnitř cévního systému a v krvi. Zdrojem tkáňového faktoru uvitř cévního systému mohou být:
- buňky z jiných tkání než z krve, které se dostanou do krve např. během porodu, při rozsáhlých poraněních, při operacích nebo s proniknutím nádorových buněk do cirkulace;
- patologické krevní buňky při myelo- a lymfoproliferačních chorobách, které mohou obsahovat ve své buněčné membráně tkáňový faktor;
- aktivované endotelie a monocyty (endotoxinem, cytokiny při systémovém zánětu), které mohou začít exprimovat ve své buněčné membráně tkáňový faktor;
- cytoplazmatický tkáňový faktor uvolněný z hemolyzovaných erytrocytů.
Při DIC se kombinuje defekt sekundární hemostázy (nízká koncentrace fibrinogenu a ostatních plazmatických koagulačních faktorů) s defektem primární hemostázy (trombocytopenie). Dochází ke krvácení z dásní (a jinde do gastrointestinálního traktu), k hematurii, může nastat epistaxe, tvoří se hematomy, krvácejí operační rány, vpichy po injekcích a jiná poranění. Může dojít ke krvácení do vnitřních orgánů včetně krvácení do mozku. Tento stav může být komplikován obliterací drobných cév mikrotromby a mikroemboly, což se může projevit periferní akrocyanózou až pregangrenózními změnami a známkami orgánových poruch (jater, ledvin, plic).
Současně s koagulací je aktivována i fibrinolýza (obr. 1.27). Ta je natolik intenzivní, že se v krvi významně zvyšuje koncentrace degradačních produktů fibrinu (Fibrin Degradation Products – FDP), k nimž patří i D-dimer. Koncentrace fibrinogenu je významně snížena. Je klinickou zkušeností, že riziko krvácení a jeho stupeň zhruba odpovídají stupni snížení koncentrace fibrinogenu.
!!!obr. 1.27.!!!
Kyselina acetylosalicylová („aspirin“) a jiné „nestroidní protizánětlivé léky“ ireverzibilně acetylují cyklooxygenázu trombocytů přítomných v krvi. Inhibují tím syntézu tromboxanu A2, který je důležitým faktorem při agregaci a degranulaci (sekreci) trombocytů vyvolaných ADP nebo adrenalinem36. Jedna dávka těchto látek (léků) proto na 5–7 dnů částečně sníží schopnost krve zacelit drobná krvácení mechanismem primární hemostázy37. Během této doby se v krvi vymění podstatná část postižených trombocytů za nové, obsahující funkční cyklooxygenázu.
!!!začátek petitu!!!
Funkce trombocytů mohou být narušeny při selhání některých orgánů, které je provázené významným narušením vnitřního prostředí. K poruše funkce trombocytů dochází při selhání renálních funkcí. Existuje přitom korelace mezi zvýšením koncentrace kreatininu nebo urey v plazmě a stupněm dysfunkce trombocytů. Krvácivé projevy mají opět příznaky poruchy primární hemostázy.
!!!konec petitu!!!
Charakter dědičnosti je autozomálně recesivní (spíše intermediární). Hemofilie C se častěji vyskytuje u etnické skupiny aškenázských Židů. Faktor XI je součástí málo významné koagulační cesty začínající aktivací faktoru XII. Klinické projevy se proto omezují na zesílení krvácení po větších poraněních nebo po operacích a u žen zesíleným menstruačním krvácením (menoragií).
Obr. 1.27 Fibrinolýza plazminem. Plazminogen je zachycen v koagulu, kde může být aktivován na proteolyticky účinný plazmin tkáňovým aktivátorem plazminogenu nebo urokinázou (proUK). Výsledkem fibrinolýzy je také vznik degradačních produktů fibrinu. Fibrinolytický účinek plazminu je tlumen inhibitorem přítomným v plazmě, který je účinný s koagulačním faktorem XIII (fibrin stabilizující faktor). Zdrojem aktivátorů plazminogenu je intaktní endotel. Ten však může být i zdrojem inhibitoru aktivátoru plazminogenu, který vzniká účinkem trombinu na endotel
36Tromboxan A2 aktivuje aktinomyozinový kompex v cytoplazmě krevních destiček.
37Inhibována je i cyklooxygenáza v jiných buňkách a tkáních. Tyto však, na rozdíl od trombocytů, mají možnost vytvoření nové funkční cyklooxygenázy, protože maji schopnost proteosyntézy a obsahují mRNA pro tento enzym. Biologický poločas účinku acetylosalicylové kyseliny je jen asi 6 hodin. Trombocyty nemají schopnost syntézy nové cyklooxygenázy, a proto jsou postiženy poruchou funkce tohoto enzymu po zbytek své přítomnosti v krvi a své existence, tj. po několik dnů.